JUNTENDO News&Events ニュース&イベント
2026.05.13 (WED)
- 順天堂大学について
- 研究活動
- 学校・塾の関係者の方へ
- メディアの方へ
- 企業・研究者の方へ
- 医学部
- 医学研究科
UFM1修飾の「基質選択の破綻」が致死的神経発達疾患を引き起こすことを解明 ― 小胞体リボソーム品質管理(ER-RQC)異常という新たな疾患メカニズム ―
順天堂大学大学院医学研究科 器官・細胞生理学 小松 雅明 主任教授らの国際共同研究グループは、UFM1修飾*1の重要因子CDK5RAP3の異常が致死的な神経発達疾患の原因となることを明らかにしました。従来のUFM1関連疾患は「酵素の異常」として理解されてきましたが、本研究は「基質選択*2の破綻」すなわちどのタンパク質にUFM1が付加されるかの異常が直接ヒト疾患を引き起こすことを初めて示しました。特に、リボソームタンパク質RPL26のUFM1修飾異常を介した小胞体リボソーム品質管理(ER-RQC)*3の破綻が、神経発達障害の分子基盤である可能性が示されました。さらに、スプライシング*4異常を標的としたアンチセンス核酸*5により分子異常が回復可能であることも示されました。本成果は、神経発達疾患の新たな理解と治療戦略の開発につながることが期待されます。
本論文はActa Neuropathologica誌のオンライン版に2026年 4月27日付で公開されました。
本研究成果のポイント
- CDK5RAP3変異による致死的神経発達疾患を世界で初めて同定
- UFM1修飾の「基質選択の破綻」と小胞体リボソーム品質管理(ER-RQC)異常が疾患の分子基盤であることを解明
- アンチセンス核酸*5によるスプライシング修復により分子異常が回復可能であることを実証
■背景
UFM1修飾はユビキチン様タンパク質による翻訳後修飾*6の一種であり、小胞体ストレス応答やタンパク質品質管理に重要な役割を担います。これまでに、UFM1やその活性化酵素UBA5、結合酵素UFC1、脱修飾酵素UFSP2に変異を持つ遺伝性神経発達疾患が報告されてきました。しかし、これらは主に酵素活性の異常に基づくものであり、「どのタンパク質がUFM1により修飾されるか」という基質選択の異常が疾患に関与するかは明らかではありませんでした。本研究は、この未解明の点に着目し、UFM1修飾の基質選択機構とヒト疾患との関連を解明することを目的として行われました。
■内容
本研究では、重度の神経発達異常を呈する2家系3例において、基質選択を担うUFM1 E3リガーゼ複合体(UFL1–UFBP1–CDK5RAP3)の構成因子であるCDK5RAP3に深部イントロン変異*7を同定しました。トリオ全ゲノム解析とRNAシーケンス解析によりスプライシング異常を検出し、原因変異を特定するとともに、トリオ全エクソーム解析データの再解析により別家系において同一変異を確認しました。さらに、遺伝子およびタンパク質レベルの解析により異常スプライシングの発生とCDK5RAP3タンパク質の著明な減少を明らかにしました。
CDK5RAP3は、リボソームタンパク質RPL26へのUFM1付加を担う基質特異性因子として機能します。本研究では、免疫沈降およびウエスタンブロット解析によりRPL26のUFM1修飾異常を確認し、その結果、小胞体におけるリボソーム品質管理機構(ER-RQC)が破綻することを明らかにしました。
さらに、プロテオミクスおよびリン酸化プロテオミクス解析により、細胞外マトリックス、細胞接着、細胞周期、神経発達に関連するシグナル経路に広範な異常が生じていることが確認されました。これらの分子異常は、変異部位を標的としたアンチセンス核酸によってスプライシングを修復することで回復可能であることも示されました。
以上の結果から、本疾患は、UFM1修飾の基質選択の破綻と、それに伴う小胞体リボソーム品質管理(ER-RQC)異常を根幹とする新たな分子病態であることが明らかになりました。
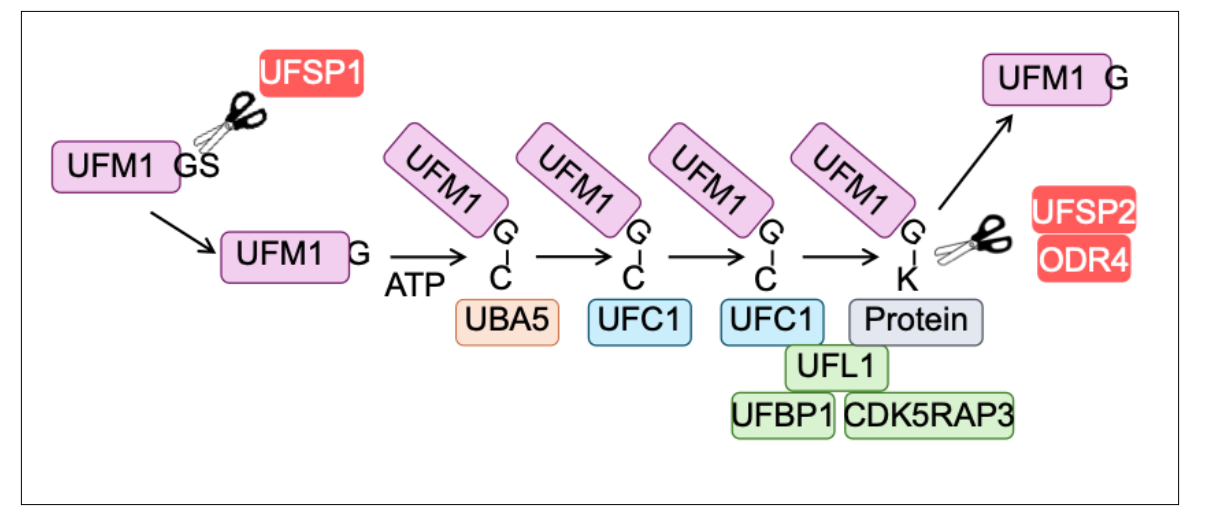
図1:UFM1修飾
UFM1修飾は、UFM1前駆体の成熟、活性化、転移、基質への付加、そして脱修飾からなる一連の反応です。まず、UFM1前駆体はUFSP1によりC末端のセリン残基(S)が切断され、グリシン残基(G)が露出します。次に、活性化酵素UBA5の活性部位システイン残基(C)にATP依存的に結合し、さらに結合酵素UFC1のシステイン残基(C)へ転移されます。その後、E3リガーゼ複合体(UFL1–UFBP1–CDK5RAP3)の働きにより、基質タンパク質のリジン残基(K)に付加されます。付加されたUFM1は、脱UFM1化酵素UFSP2によって取り除かれます。これまでに、UFM1、UBA5、UFC1、UFSP2に変異を持つ患者で重篤な遺伝性小児神経発達疾患が報告されており、本経路が神経発達に不可欠であることが示されてきました。本研究では、基質選択を担うE3リガーゼ複合体の構成因子CDK5RAP3の異常も同様に疾患を引き起こすことを明らかにし、UFM1修飾経路全体の破綻が神経発達障害の原因となることを示しました。
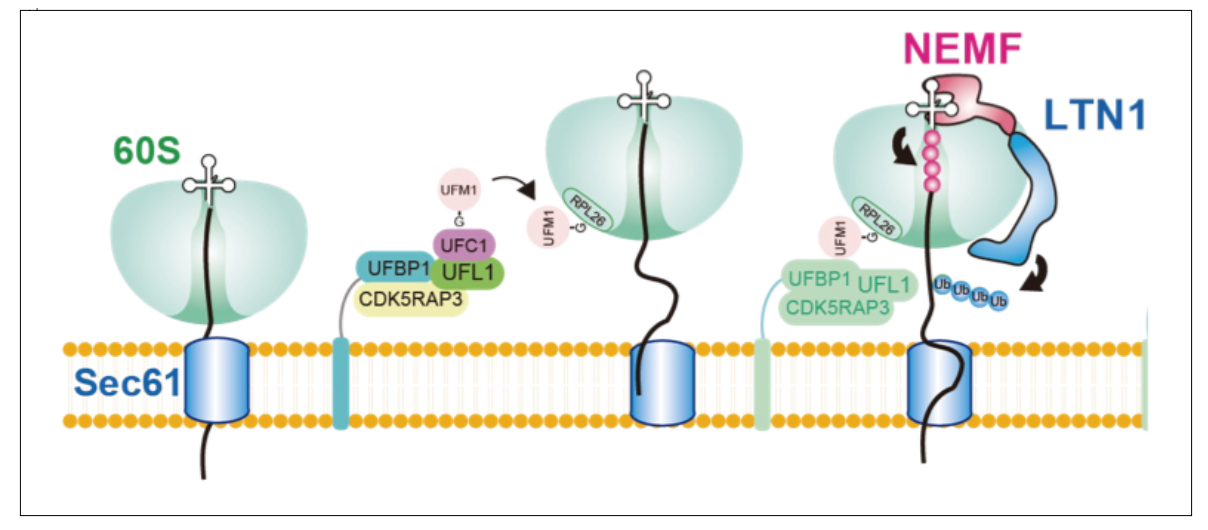
図2:UFM1 E3リガーゼによる基質選択とERリボソーム品質管理(ER-RQC)の模式図
小胞体膜上のSec61トランスロコンに結合したリボソームにおいて翻訳異常が生じた際、UFM1 E3リガーゼ複合体(UFL1–UFBP1–CDK5RAP3)はリボソームタンパク質RPL26を基質として選択的にUFM1修飾を付加します。この修飾は、小胞体におけるリボソーム品質管理機構(ER-RQC)に関与し、LTN1やNEMFなどのRQC因子による新生鎖タンパク質のユビキチン化と分解を介した異常翻訳産物の除去を促進します。本研究では、CDK5RAP3変異によりこの基質選択が障害されることでER-RQCが破綻することを明らかにし、UFM1 E3リガーゼ複合体による基質選択が本過程に重要であることを示しました。
■今後の展開
本研究は、UFM1修飾異常疾患の理解を「酵素活性の異常」から「基質選択の異常」へ拡張しました。特に、ER-RQCという翻訳品質管理機構の破綻が神経発達疾患の根底にある可能性を示した点は、今後の神経疾患研究に大きな影響を与えると考えられます。
また、深部イントロン変異によるスプライシング異常という新たな病因メカニズムを提示するとともに、アンチセンス核酸による修復の可能性を示したことから、個別化医療への応用が期待されます。今後は神経系モデルを用いた検証や臨床応用に向けた研究が重要となります。
■用語解説
*1 UFM1修飾: 翻訳後修飾の一種で、ユビキチン様タンパク質UFM1が標的タンパク質に結合することで、細胞内の品質管理やストレス応答を制御する仕組み。
*2 基質選択: どのタンパク質に修飾が付加されるかを決定する仕組み。
*3 小胞体リボソーム品質管理(ER-RQC): 翻訳異常を検知し、リボソームを制御する品質管理機構。
*4 スプライシング: RNAから不要部分を除去し正しいタンパク質情報を作る過程。
*5 アンチセンス核酸: RNAに結合して遺伝子発現やスプライシングを制御する分子。
*6 翻訳後修飾(Post-translational modification): タンパク質が細胞内で作られた後に、化学的な修飾(例:リン酸化、ユビキチン化など)を受けることで、その機能や安定性、細胞内での働きが調節される仕組み。
*7 深部イントロン変異: イントロン内部に生じる変異で、異常なスプライシングの原因となる。
■原著論文
本研究はActa Neuropathologica誌のオンライン版に2026年4月27日付で公開されました。
タイトル: A severe neurodevelopmental syndrome linked to a South Asian founder variant in the UFMylation adaptor CDK5RAP3
タイトル(日本語訳): 南アジアに広がる共通変異が引き起こす重い神経発達障害 ― CDK5RAP3の異常が原因 ―
著者: Michaela Yuen1,2,3, Katharine Zhang1,2,3, Rhett G. Marchant1,2,3, 石村 亮輔4, Mark Graham5, May Aung-Htut6,7, Samantha Bryen8,9, Rocio Rius8,9,10, Lee Marshall11, Nader Aryamanesh3,11, Gregory Dziaduch1,2,3, Himanshu Joshi1,2,3, Steve D. Wilton6,7, Meredith Wilson3,12, Russell Gear13,14, Helen Doyle15, Michael Krivanek15, Richard J. Leventer13,16,17, Susan M. White10,14,18, Sarah A. Sandaradura12,13, 小松 雅明4, Frances J. Evesson1,2,3*, Sandra T. Cooper1,2,3*
著者所属: 1)Kids Neuroscience Centre, Kids Research, The Children’s Hospital at Westmead, NSW, Australia, 2)The Children’s Medical Research Institute, Westmead, NSW, Australia, 3)School of Medical Sciences, Faculty of Medicine and Health, The University of Sydney, Australia, 4)順天堂大学大学院医学研究科器官・細胞生理学, 5)Biomedical Proteomics, Children’s Medical Research Institute, Australia, 6)Perron Institute for Neurological and Translational Science, Australia, 7)Murdoch University, Health Futures Institute, Australia, 8)Centre for Population Genomics, Garvan Institute of Medical Research, UNSW Sydney, Australia, 9)Murdoch Children’s Research Institute, Australia, 10)Department of Paediatrics, University of Melbourne, Australia, 11)Bioinformatics Group, Children’s Medical Research Institute, Australia, 12)Department of Paediatrics, University of Sydney, Australia, 13)Department of Neurology, The Royal Children’s Hospital Melbourne, Australia, 14)Victorian Clinical Genetics Services, Australia, 15)Department of Histopathology, The Children’s Hospital at Westmead, Australia, 16)Department of Paediatrics, University of Melbourne, Australia, 17)Murdoch Children’s Research Institute, Australia, 18)The Mercy Hospital for Women, Australia
DOI: https://doi.org/10.1007/s00401-026-03017-2