Department of Research for Parkinson's Disease
Research
Click on the title to view the contents
Hereditary Parkinson's Disease
Hereditary forms of Parkinson's disease are found in 5-10% of cases. Recently, the causative genes of Parkinson's disease have been identified, and a molecular biological approach has become possible (Table 1). Mutations in these genes cause degeneration of the dopaminergic nerves in the substantia nigra of the midbrain, leading to Parkinson's disease, but it is not clear how mutations in each gene product contribute to neurodegeneration or whether there is a common neurodegenerative mechanism.
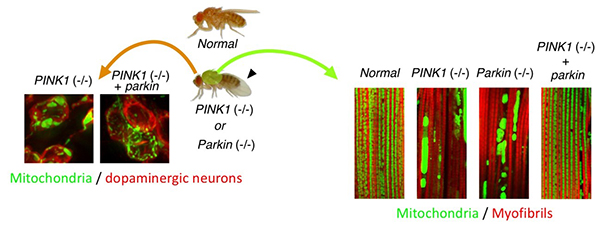
Figure 1. Drosophila Parkinson's disease
models by PINK1 and Parkin mutations show mitochondrial degeneration.
Drosophila also possesses the PINK1 and Parkin genes, and these knockout (-/-)
Drosophila show degeneration of mitochondria (green in the photos) in muscles (right) and
dopaminergic neurons (left), and drooping wings (arrowhead) (Imai, PLoS
Genet. 2010). When the Parkin gene is exogenously introduced into PINK1
knockout Drosophila, the mitochondrial degeneration is improved (far right in each
photo).
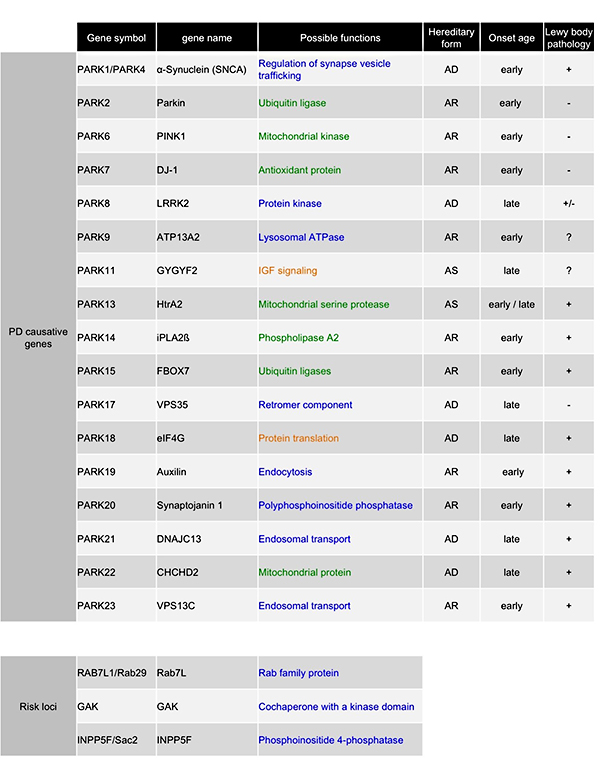
Table 1. Causative genes of Parkinson's disease.
AD; autosomal dominant inheritance, AR; autosomal recessive inheritance, AS; susceptibility
gene. Variants of two genes (DNAJC13 and TMEM230) have been found in PARK21
from the same family, and it is unclear which is the etiologic gene. Blue indicates genes
related to vesicular trafficking, green indicates genes related to mitochondria, and orange
indicates genes with other functions. These gene products are proteins with various functions,
but whether they function independently or in a common pathological pathway is an important
question to be answered. We have shown that there is a genetic interaction between Parkin and
PINK1 using Drosophila as a model (Yang, PNAS 2006).
Drosophila models of Parkinson's disease
To clarify this issue, we are using Drosophila ,
which has a short lifespan suitable for molecular genetic analysis (Figs.
1, 5, 6). Drosophila has
dopaminergic neurons and ages in the same way as humans. For example, motor and cognitive
abilities decline, lipid oxidation occurs, and reproductive abilities decline. It takes several
years to clarify the relationship between multiple Parkinson's disease genes using mice as a
model, but with Drosophila , the relationship can be determined within a year.
We are using the model fly to clarify the functions of the Parkinson's disease-causing genes
related to mitochondria (Fig. 7) and vesicular trafficking (Figs. 8-12) as shown in Table 1 and the mechanism
of neurodegeneration caused by their mutations.
Proteomics Analysis
Molecular genetic analysis alone does not reveal the
functions of the proteins produced by the genes responsible for Parkinson's disease. We are
studying how the etiological mutations of Parkinson's disease affect the function of proteins by
culturing human cultured cells and primary cultured neurons and by proteomic analysis. We also
use mouse models of Parkinson's disease to analyze the function of proteins. The roles of
Parkinson's disease-causing genes
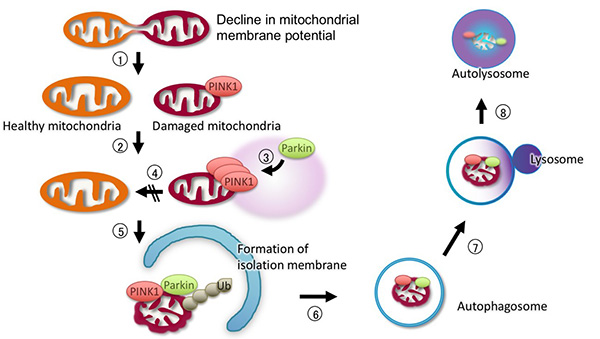
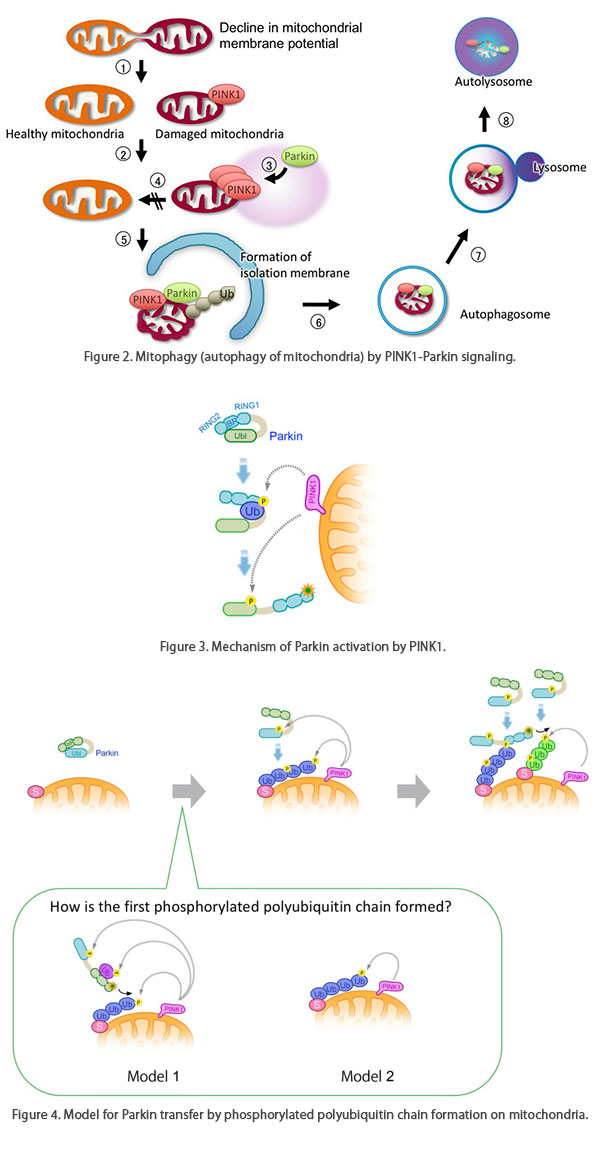
Figure 2. Mitophagy (autophagy of mitochondria) by
PINK1-Parkin signaling.
PINK1-Parkin signaling removes defective mitochondria through the following steps.
(1) Defective mitochondria with reduced membrane potential are fragmented, and (2) they do not
re-fuse. (2) PINK1 accumulates on the mitochondria with reduced membrane potential, and kinase
activity is activated. (3) PINK1 activates Parkin, and Parkin is transferred from the cytoplasm
to the mitochondria. Next, Parkin ubiquitinates mitochondrial outer membrane proteins (Ub).
Autophagy-related proteins such as TBK1, Optineurin, and LC3 are recruited to the ubiquitinated
mitochondria, and the defective mitochondria are degraded by the autophagic pathway.
Figure 3. Mechanism of Parkin activation by PINK1.
Under steady state conditions, Parkin is compactly folded in the cytoplasm as an inactive state.
When the membrane potential is decreased by mitochondrial dysfunction, PINK1 is activated.
Ubiquitin (Ub), which is phosphorylated (Ⓟ) by activated PINK1, enters the RING1-IBR
region of Parkin, which loosens the Parkin structure, and then PINK1 phosphorylates the
ubiquitin-like region (Ubl) of Parkin. This exposes the active center of the ubiquitin ligase,
and Parkin becomes an active ubiquitin ligase.
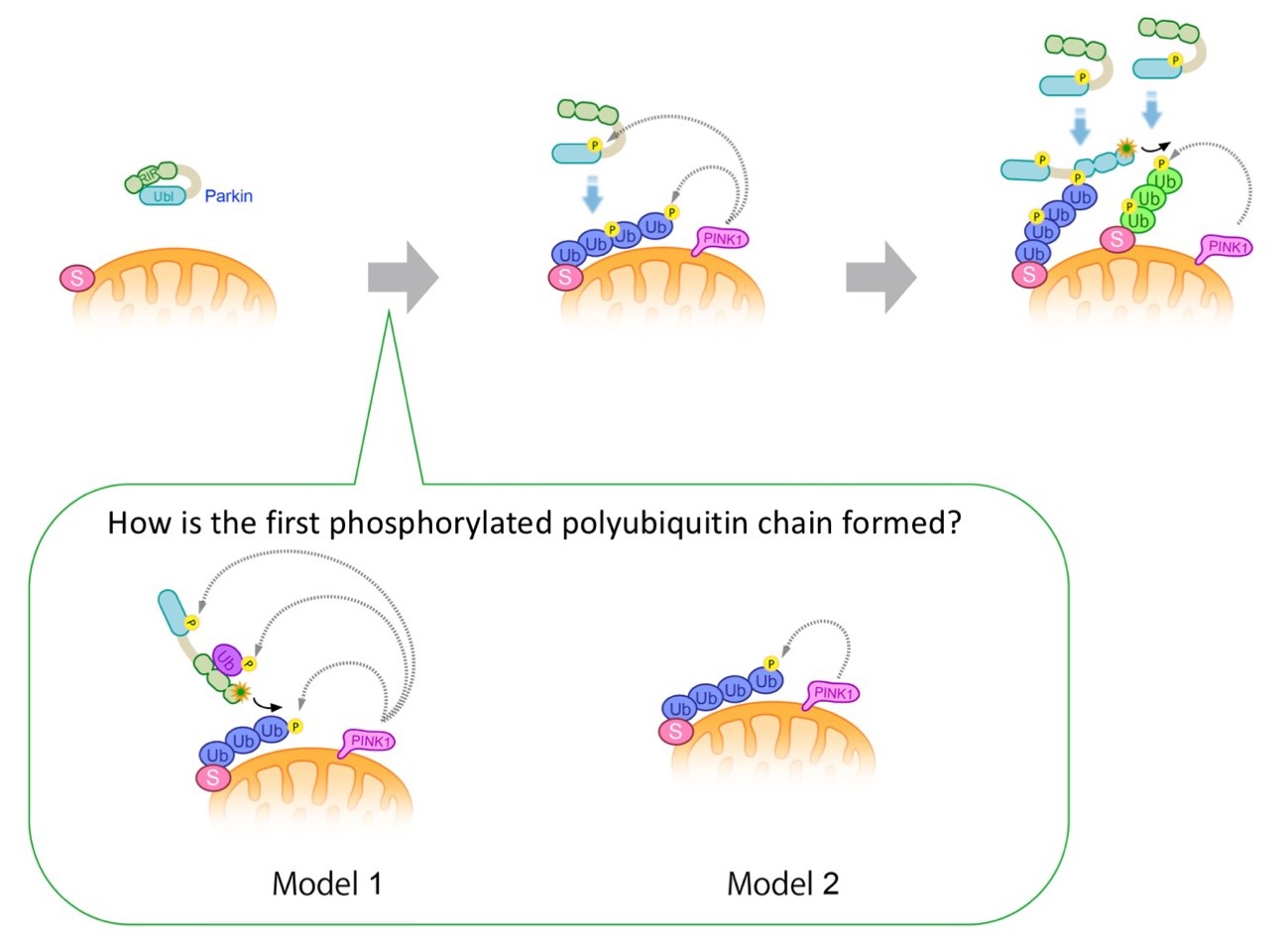
Figure 4. Model for Parkin transfer by
phosphorylated polyubiquitin chain formation on mitochondria.
Parkin localizes in the cytoplasm as an inactive ubiquitin ligase (left).
When mitochondria are damaged and the membrane potential is decreased, PINK1 accumulates and
activates, phosphorylating polyubiquitin chains on the mitochondira and the ubiquitin-like
domain of Parkin. Parkin has an affinity for phosphorylated polyubiquitin chains and localizes
to mitochondria (center).
Binding of Parkin to the phosphorylated polyubiquitin chain activates ubiquitin ligase activity,
which further generates the polyubiquitin chain to mitochondrial outer membrane proteins.
Morover, phosphorylation of this polyubiquitin chain by PINK1 leads to amplification of the
phosphorylated polyubiquitin chain on the mitochondrial outer membrane, achieving rapid
mitochondrial transfer and activation of Parkin (right).
Ub; ubiquitin, P; phosphorylation, S; ubiquitinated substrate protein on the mitochondrial outer
membrane.
(bottom box) How is the first phosphorylated polyubiquitin chain formed?
Model 1: Parkin is activated by mono-ubiquitin that is phosphorylated by PINK1.
There is a possibility that activated Parkin ubiquitinates substrates on mitochondria by free
diffusion, which is phosphorylated by PINK1.
Model 2: PINK1 phosphorylates the ubiquitin chains of mitochondrial outer
membrane proteins that are physiologically modified by ubiquitin ligases other than Parkin,
which could be used as the initial scaffold for Parkin localization to mitochondria. We prefer
the Model 2 (Shiba-Fukushima, PLoS Genet. 2014b; figures reproduced from
Imai et al. Experimental Medicine 2014).
Research with clinical samples
In cooperation with the Department of Neurology at Juntendo University, we also use iPS cells established from Parkinson's disease patients. The purpose of this is to confirm whether what we have learned from Drosophila and proteomic analysis is happening in the patient's body. For example, one of the ubiquitinated substrates of Parkin that we found in the Drosophila model is Miro, which is a protein necessary for mitochondrial transport, and when Parkin is activated, mitochondrial transport in neuronal axons arrests (Fig. 5). The events observed in flies were also reproduced in dopaminergic neurons generated from human iPS cells (Fig. 6). Furthermore, the transport of defective mitochondria was less likely to stop in the dopaminergic nerves of patients with mutations in Parkin (Shiba-Fukushima, Hum Mol Genet. 2017).
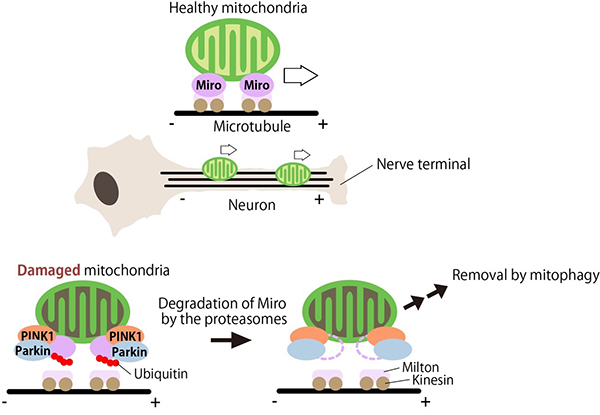
Figure 5. Stability regulation of Miro by
PINK1-Parkin contributes to the mitochondrial quality control in neurons.
Miro is responsible for microtubule transport of mitochondria. In neurons, Miro carries
mitochondria to nerve terminals. In damaged mitochondria, PINK1 activates Parkin, which degrades
Miro. This prevents the transport of damaged mitochondria to the nerve terminals. Damaged
mitochondria that are retained in the cell body are thought to be degraded by mitophagy (Liu,
PLoS Genet. 2011).
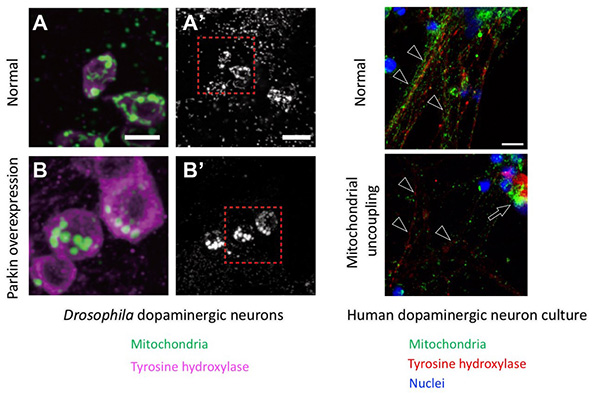
Figure 6. Mitochondrial transport in dopaminergic
neurons by PINK1-Parkin.
(A-B') Dopaminergic neurons (tyrosine hydroxylase-positive cells) in the fly brain. The image on
the left is a magnified view of the red box on the right. (A', B') Mitochondrial morphology in
dopaminergic nerves. (A, A') Mitochondrial image of a normal dopaminergic neuron. (B, B')
Overexpression of Parkin results in the degradation of Miro and the loss of mitochondrial
signals in nerve terminals. On the other hand, fragmented mitochondria accumulate in the
neuronal cell bodies (Shiba-Fukushima, PLoS Genet. 2014b). In
dopaminergic neurons differentiated from human iPS cells, mitochondria in nerve axons
(arrowheads) disappear and accumulate in the cell bodies (arrows) after treatment for the
membrane potential reduction, as in flies. Scales: 5 µm (A, B), 10 µm (A', B'), 10 µm (human
neurons).
Regulation of mitochondrial function by Parkinson's disease-causing genes
Mitochondria, one of the organelles of the cell, have various functions such as ATP synthesis, which is the source of energy for the body, lipid metabolism, iron metabolism, regulation of intracellular Ca2+ concentration, and regulation of cell death signals. Genetic evidence has revealed that mitochondrial dysfunction or dysregulation are involved in neurodegenerative diseases such as Parkinson's disease (Fig. 7, Table 1), amyotrophic lateral sclerosis (ALS), and frontotemporal dementia (FTD)-ALS. For example, in Parkinson's disease, the aforementioned juvenile Parkinson's disease genes PINK1 and Parkin have been shown to be involved in mitochondrial quality control (removal of broken mitochondria). On the other hand, the late-onset Parkinson's disease gene CHCHD2 regulates the electron flow in the mitochondrial respiratory chain complex (Meng, Nat Commun. 2017), and it has been found that when Parkinson's disease mutations are introduced in CHCHD2, electrons leak out, leading to oxidative stress.
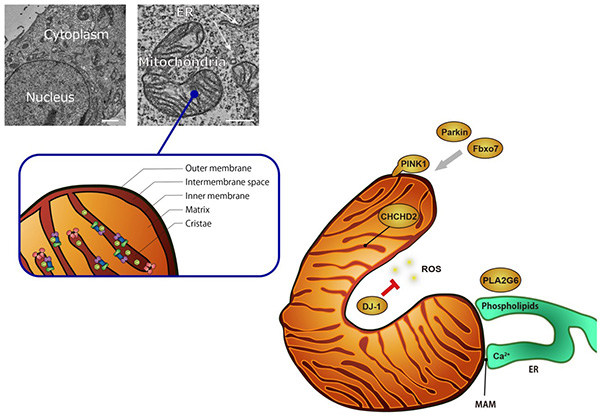
Figure 7. Possible functions of Parkinson's
disease gene products in mitochondria.
PINK1-Parkin is involved in mitochondrial
quality control; Fbxo7 also works with Parkin; DJ-1 is involved in the removal of reactive
oxygen species (ROS) generated from mitochondria; CHCHD2 is present in the mitochondrial lumen
and is involved in maintaining the activity of the respiratory chain complex; PLA2G6 is involved
in the removal of lipids peroxidized by ROS generated in mitochondria and the regulation of
Ca2+ influx from the endoplasmic reticulum (ER) through phospholipid remodeling.
Mitochondria exchange substances (Ca2+, lipids, etc.) with the ER, and the proximal
region (less than 30 nm) is called the Mitochondria Associated Membrane (MAM), which is
connected by a protein complex. Scale in electron micrographs: 2 μm (left), 500 nm (right).
(Figure from Imai Y, 日本臨牀, 2017)
Regulation of vesicular trafficking by Parkinson's disease-causing genes
Vesicular transport is an intracellular phenomenon in
which substances are transported in a closed lipid membrane. It is used for the exchange of
substances between cellular organelles (mitochondria, endoplasmic reticulum, Golgi apparatus,
lysosomes, etc.). It is also used for the release (exocytosis) and uptake (endocytosis) of
substances outside the cell.
It has become clear that many of the genes responsible for Parkinson's disease are involved in
intracellular vesicle trafficking (Figs. 8, 9). These
genes have been found to function in both neuronal and non-neuronal cells. In particular, we and
other researchers have shown that they are involved in endocytosis from presynapses (Fig. 10) in dopaminergic neurons, which is closely related to the
pathology of Parkinson's disease (Inoshita T, Hum Mol Genet. 2017,
Inoshita T, J Genet. 2018).
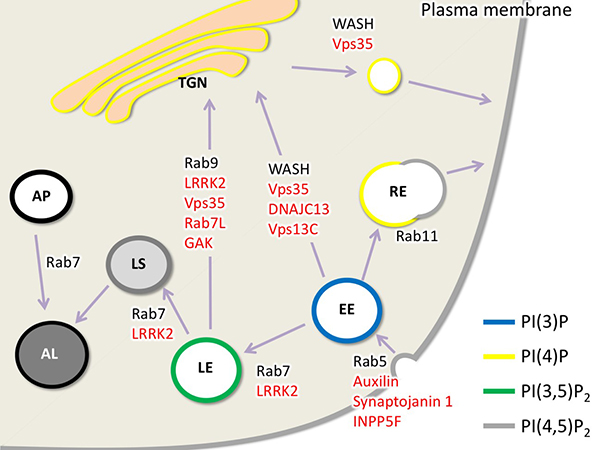
Figure 8. Functional diagram of the Parkinson's
disease gene product assumed in vesicular transport.
Here, vesicular trafficking from the plasma membrane to endocytosis, endosomes, lysosomes, and
the Golgi apparatus is shown. Red genes indicate the genes responsible for Parkinson's disease
or susceptibility genes. Each vesicle is surrounded by inositol phospholipids with special
phosphorylation modifications: TGN, trans-Golgi network; AL, autolysosome; AP, autophagosome;
LS, lysosome; EE, early endosome; LE, late endosome; RE, recycling endosome. (Figure from
Inoshita and Imai, AIMS Mol Sci. 2015)
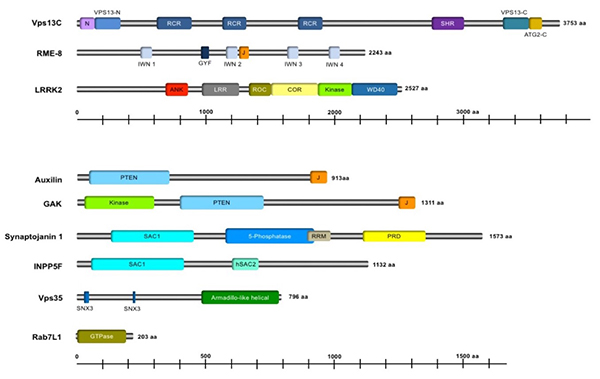
Figure 9. Domain structure of Parkinson's disease
gene products involved in membrane transport.
For details of the domains, please refer to the cited reference (figure reproduced from Inoshita
T, et al., J Genet. 2018, Figure 4).
Synaptic endocytosis
Neurons exchange signals via synapses (Fig. 11). A synapse is a junction between a nerve and a nerve or muscle fiber or other cells. Signals are exchanged with neurotransmitters and electrical signals. Neurotransmitters are released by vesicles (specifically called synaptic vesicles) from the neurons that carry the signal. After the vesicles fuse with the synaptic membrane, they are retrieved by endocytosis. The Drosophila neuromuscular synapse is well suited for studying the uptake of synaptic vesicles and the abnormalities caused by mutations in the gene responsible for Parkinson's disease (Fig. 12).
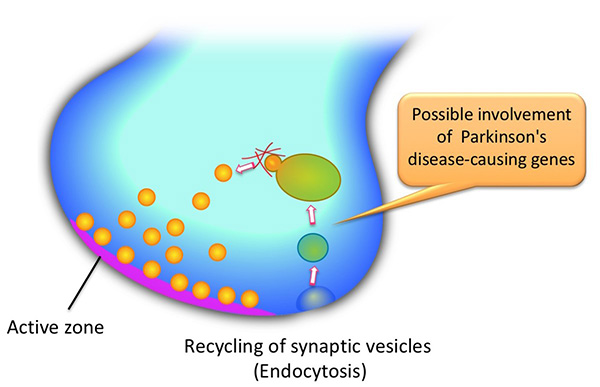
Figure 10. Possible participation parts of
Parkinson's disease gene products in neurons.
At least two or more of the Parkinson's disease-causing genes involved in vesicular trafficking
may be involved in presynaptic endocytosis (retrieval of synaptic vesicle membranes) and
regeneration of synaptic vesicles. The relationship between the genes can be clarified by
Drosophila molecular genetics.
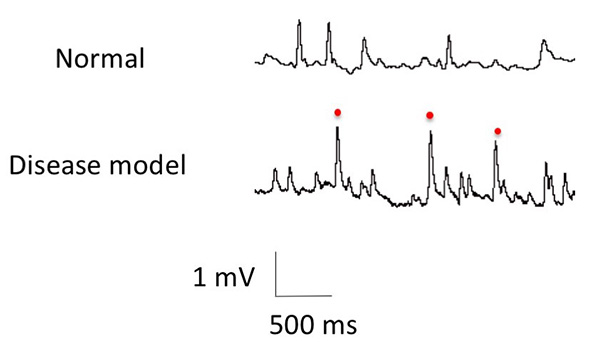
Figure 11. Altered synaptic functions caused by
the genes responsible for Parkinson's disease.
Electrical signals at the neuromuscular junctions in Drosophila . The pulses show the
signals from the neurons to the muscle. Abnormal signals (indicated by red dots) can be seen in
Drosophila with Parkinson's disease mutations.
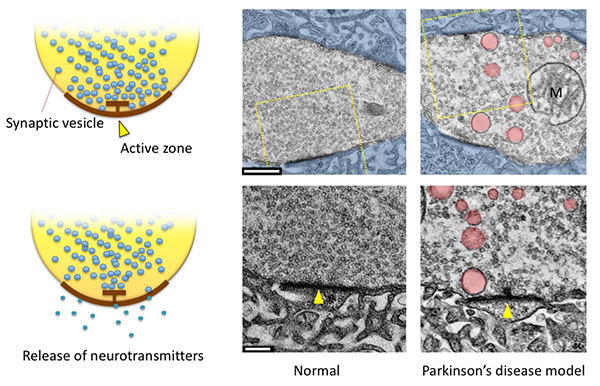
Figure 12. Synapses at the neuromuscular junctions
in Drosophila larvae.
(Left) Synaptic vesicles dock with the presynaptic membrane structure, called the active zone,
and release neurotransmitters stored inside.
(Right) Electron micrographs of synapses. (Top) Presynapses (motor nerve terminals) are
uncolored, postsynapses (muscle cells in this case) are shown in blue. (Bottom) Magnified view
of the yellow dashed area. Active zones are indicated by arrowheads. Abnormally large vesicles
(pink) are seen at presynapses in a Parkinson's disease model. The smaller vesicles are synaptic
vesicles with a diameter of about 35 nm; scale: 500 nm (top), 200 nm (bottom).
The Parkinson's disease-causing gene is thought to be involved in the process from endocytosis
to regeneration of synaptic vesicles.
Is Parkinson's disease a prion disease?
It is thought that α-Synuclein repeatedly binds to and
releases from synaptic vesicles during synaptic exocytosis (Fig. 13). In Parkinson's disease, it
is becoming clear that α-Synuclein garbage (fibrils and inclusions) accumulates in the brain and
leads to neurodegeneration (Fig. 14). It has been experimentally
confirmed that this α-synuclein garbage spreads through the brain via neural circuits, just as
mold from rotten oranges spreads to other oranges (Fig. 15). The research
question “What are the conditions (genes, neural activity) that increase the risk of spreading
the disease?” (Fig. 16) is important to prevent the onset of Parkinson's
disease.
We have developed a Parkinson's disease model fly that reproduces α-Synuclein garbage, and have
identified the conditions that increase the risk of garbage spreading (Fig.
17). The Parkinson's disease-causing gene PLA2G6/iPLA2β (see Table 1) encodes a
phospholipase. When disease-associated mutations are introduced to this enzyme, the phospholipid
membrane becomes thinner with age. As a result, the size of the synaptic vesicles to which
α-Synuclein binds becomes smaller, and α-Synuclein is more likely to be released into the
cytoplasm. This is thought to be a risk for α-Synuclein aggregation (Mori,
PNAS 2019).
Currently, an efficient method to detect α-Synuclein garbage (disease-causing structures and
aggregated α-Synuclein) has been developed. Using this method, we are exploring strategies to
prevent the formation of α-Synuclein garbage in humans and flies (Figure
18).
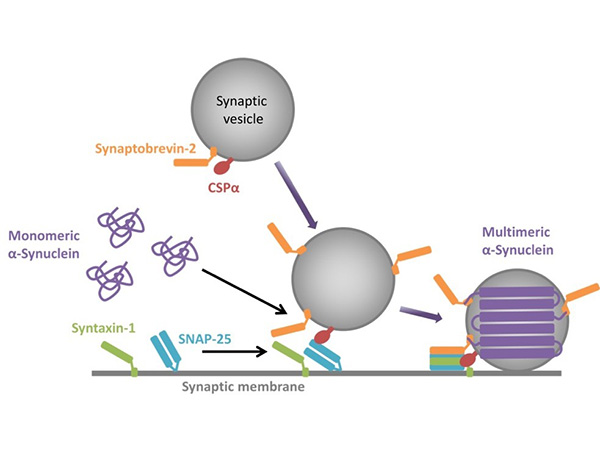
Figure 13. α-Synuclein undergoes repeated
multimerization and disassembly during synaptic vesicle secretion.
When a synaptic vesicle docks to the synaptic membrane, α-Synuclein polymerizes on the membrane
(Multimeric α-Synuclein). Upon completion of secretion, α-Synuclein detaches from the synaptic
vesicle membrane and returns to its monomeric form (Monomeric α-Synuclein). During neural
activity, α-Synuclein is thought to undergo this repeated polymerization and disassociation. At
some point, α-Synuclein undergoes a conformational change and becomes fibrils (Figure reproduced
from Inoshita T, et al., J Genet. 2018, Figure
5).
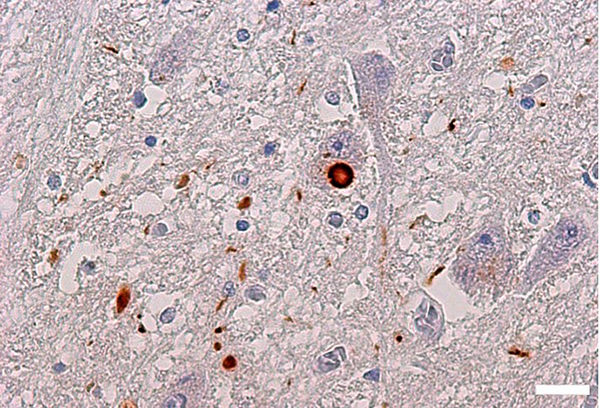
Figure 14. α-Synuclein inclusions (Lewy bodies) in
Parkinson's disease brain.
Brown-colored aggregates are α-Synuclein inclusions. The brown spheres in the center are Lewy
bodies found in neuronal cell bodies, which are one of the markers for pathological diagnosis of
Parkinson's disease. Parkinson's disease brain (pons) stained with α-Synuclein fibril-specific
antibody. Scale: 20 μm.
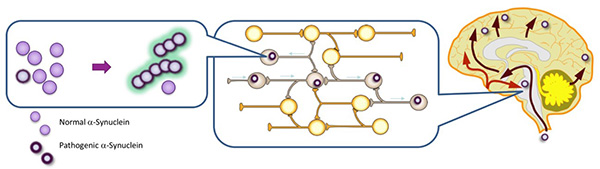
Figure 15. Pathologically structured α-Synuclein
is transmitted like a prion.
α-Synuclein with pathological structural changes in neurons propagates between neurons and
amplifies the pathological α-Synuclein.
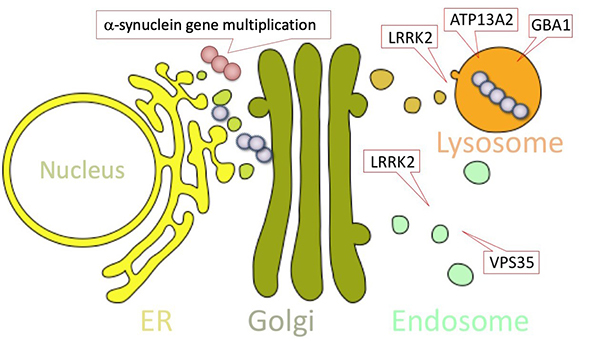
Figure 16. Parkinson's disease-causing genes that
contribute to the amplification of α-Synuclein in pathological structures.
Duplication of α-Synuclein gene: normal cells have one pair (two copies) of α-Synuclein
gene, but gene duplication increases the amount of α-Synuclein protein produced, which
contributes to the accumulation and propagation of α-Synuclein in pathogenic structures.
Mutations in genes involved in intracellular transport and garbage disposal: Parkinson's
disease-causing genes such as LRRK2, VPS35, and ATP13A2 are thought to be
involved in protein transport and garbage disposal. Mutations in these genes prevent the
degradation of pathogenic α-synuclein, and lead to Parkinson's disease. (Figure reprinted from
Imai Y, Clinical Practice of Brain and Nerve Diseases, Parkinson's Disease and Movement
Abnormalities, Nakayama Shoten, 2013)
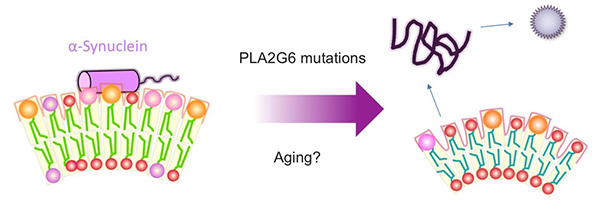
Figure 17. Mechanism of α-Synuclein aggregation
caused by mutations in the Parkinson's disease-causing gene PLA2G6
When PLA2G6 is mutated, the size of the synaptic vesicle is reduced. The change causes the
membrane to become more curved, making it easier for α-synuclein to detach from the synaptic
vesicle membrane, which may be a risk for aggregation.
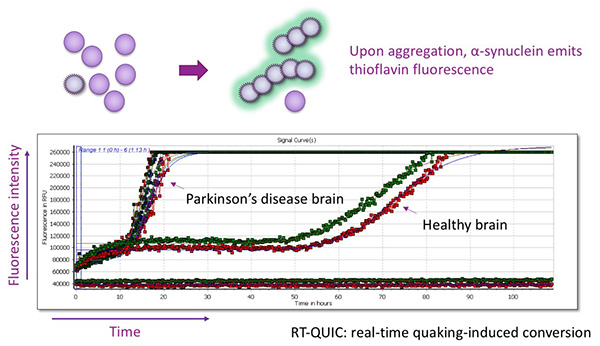
Figure 18. RT-QUIC method for detecting
α-Synuclein with pathogenic structure.
Using the property that α-Synuclein in the pathogenic structure converts normal α-Synuclein into
the pathogenic structure and amplifies it, the amount of α-Synuclein in the pathogenic structure
is evaluated. The higher the amount of pathogenic α-Synuclein, the faster the fluorescence
signal increases.
Mitochondria and α-Synuclein
It has been reported that mitochondrial function is
impaired in Parkinson's disease. PINK1 and Parkin, which are related to mitochondrial quality
control, and CHCHD2, which regulates electron transfer in the mitochondrial respiratory chain
complex, have also been reported as Parkinson's disease-causing genes. α-Synuclein aggregation
is also linked to the development of Parkinson's disease. However, the relationship between
α-Synuclein aggregation and mitochondria has been unclear.
We found widespread presence of α-Synuclein inclusions (Lewy bodies) in the brains of
Parkinson's disease patients with CHCHD2 mutations (Fig. 19).
Moreover, dopaminergic neurons derived from iPS cells generated from patients with
CHCHD2 mutations and Drosophila with CHCHD2 mutations were also found
to accumulate α-Synuclein inclusions (Ikeda, Hum Mol Genet. 2019).
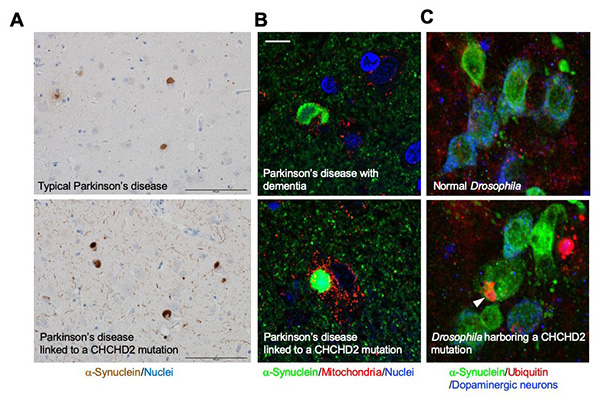
Figure 19. Mutations in the mitochondrial protein
CHCHD2 lead to the aggregation of α-Synuclein.
(A) A Parkinson's disease patient with a CHCHD2 mutation show extensive accumulation of
α-synuclein in the brain (bottom). The accumulation is more pronounced than in patients with
sporadic Parkinson's disease (top). The round, brown-colored objects are Lewy bodies (see also
Fig. 14). Scale: 100 μm. (B) Higher magnification view of Lewy bodies
(round green structures) in a Parkinson's disease patient with a CHCHD2 mutation
(bottom). Lewy bodies are little co-localized in mitochondria (red). Extensive Lewy bodies are
also seen in Parkinson's disease patients with dementia (top), but there is no significant
difference in property when compared to these Lewy bodies. Scale: 10 μm. (C) Dopaminergic
neurons in Drosophila with a CHCHD2 mutation, showing aggregation of α-synuclein
(green) and accumulation of ubiquitin (red, an arrowhead). Ubiquitin, a molecule responsible for
degradation, is thought to accumulate together with abnormal proteins. It is known that Lewy
bodies also contain ubiquitin.
Delivering hydrogen ions to mitochondria!?
As noted above, mutations in the CHCHD2 gene
result in reduced mitochondrial function and accumulation of α-Synuclein inclusions (Ikeda,
Hum Mol Genet., 2019). Drosophila with mutations in the
CHCHD2 gene produces large amounts of reactive oxygen species from their mitochondria
(Meng, Nat Commun., 2017) and accumulates α-Synuclein inclusions
(Ikeda, Hum Mol Genet., 2019).
Hydrogen ions (protons) have the ability to remove reactive oxygen species. Therefore, we tried
to remove reactive oxygen species by generating hydrogen ions in mitochondria. In detail, we
introduced a protein called Delta-rhodopsin, which is found in archaea, into the mitochondria
(Fig. 20). This Delta-rhodopsin has the property of transporting hydrogen
ions when exposed to light. By taking advantage of this property of Delta-rhodopsin, we made it
possible to transport hydrogen ions to the outside of mitochondria when exposed to light. The
hydrogen ions gathered on the outside remove reactive oxygen species and also drive the
mitochondrial energy-producing machinery.
The idea worked, energizing the mitochondria of a Drosophila model of Parkinson's
disease with a CHCHD2 mutation, and also reducing neurodegeneration (Imai, Commun
Biol. 2019). Surprisingly, we also found that α-Synuclein inclusions were
no longer accumulated (Fig. 21). This observation indicates that
mitochondria are capable of actively removing protein garbage. We are currently conducting
research to clarify this mechanism of mitochondria and to apply it to preventive methods for
Parkinson's disease.
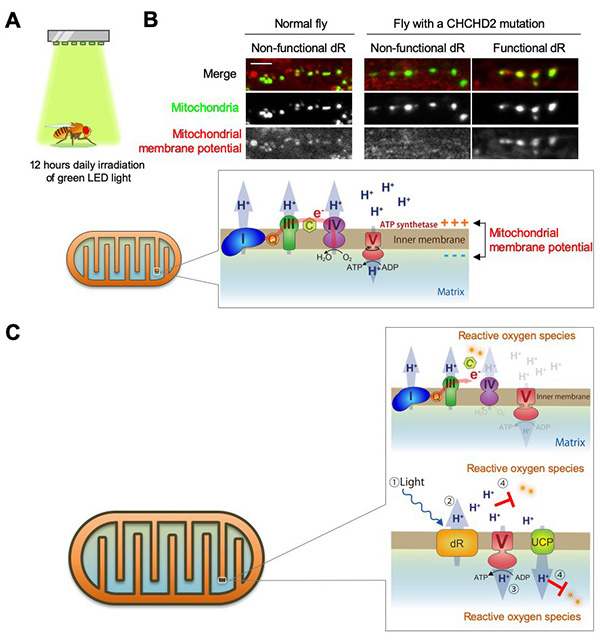
Figure 20. Delta-rhodopsin delivers hydrogen ions
to mitochondria.
(A) Light irradiation of CHCHD2 mutant flies with Delta-rhodopsin (dR) introduced into
the mitochondrial inner membrane. Flies do not have a skull, so green light reaches deep into
the brain. (B) Mitochondrial membrane potential of CHCHD2 mutant flies is restored by
dR transduction. Mitochondrial membrane potential is monitored with a reagent called TMRM. A dR
mutant that does not function when exposed to light was placed as a comparison control. In
healthy mitochondria, the respiratory chain complexes I, III, and IV pump hydrogen ions
(protons) out of the matrix using sugar from the diet as material. Complex V then returns the
pumped protons to the matrix side, where energy (ATP) is produced. Thus, in healthy
mitochondria, the membrane potential (the bias between + and - across the membrane; about -150
mV) is maintained. Scale: 10 μm. (C) When CHCHD2 is broken, electrons flowing through
the respiratory chain complexes I, III, and IV leak, producing reactive oxygen species. Since
electrons leak out, protons cannot be pumped out efficiently, resulting in a decrease in the
membrane potential (picture above in the balloon). dR pumps out protons instead of the
respiratory chain complexes I, III, and IV when exposed to light. Protons also have the ability
to remove reactive oxygen species. Uncoupling protein (UCP) is a protein that produces body heat
while returning protons to the matrix side, and the protons returned to the matrix side by UCP
also remove reactive oxygen species from the matrix side (see picture below in the balloon). For
the overall structure of the mitochondria, see Figure 7.
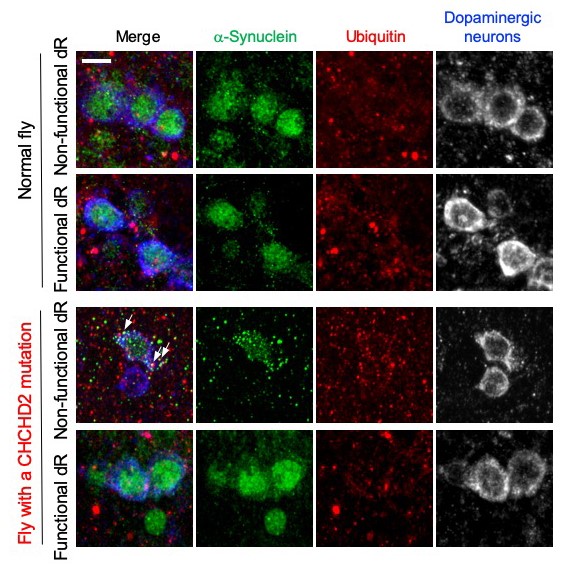
Figure 21. Mitochondria activated by
Delta-rhodopsin prevents α-synuclein aggregation.
Normal and CHCHD2 mutant flies expressing Delta-rhodopsin (dR) were exposed to light. In the
CHCHD2 mutant flies with non-functional dR, which does not have the ability to pump out protons,
α-synuclein aggregates appear (third row, green granules). These aggregations partially
co-localize with the granular signal (red) of ubiquitin (arrow). When functional dRs were
introduced into CHCHD2 mutant flies, the aggregation of α-synuclein was suppressed, as in normal
flies (fourth row). Scale: 5 μm.
Search for Parkinson's disease drugs by combining iPS cells and fly models
As noted above, when mitochondria are dysfunctional, α-Synuclein inclusions accumulate (Ikeda, Hum Mol Genet. 2019). Conversely, when mitochondria are energized with Delta-rhodopsin, α-Synuclein inclusions are eliminated (Imai, Commun Biol. 2019). This indicates that drugs to improve mitochondrial functions are promising drugs for Parkinson's disease prevention. PINK1 and Parkin, described in the section on Proteomics Analysis, are responsible for mitochondrial quality control. In other words, they are thought to be sorting out mitochondria that have lost their function and keep healthy mitochondria in the cells. Parkin is an enzyme involved in protein degradation called ubiquitin ligase. However, mutated Parkin in patients is thought to not work properly even though mitochondrial function is impaired. Therefore, we collaborated with researchers at Takeda Pharmaceutical Company to search for drugs that can activate Parkin, and found two candidate drugs (Shiba-Fukushima, iScience 2020). To evaluate the drugs, we used a fly model of Parkinson's disease with reduced activity of PINK1 (a mitochondrial kinase that activates Parkin) and dopaminergic neurons generated from iPS cells to reduce the cost and speed up the evaluation process (Fig. 22). Currently, we are studying the mechanism by which the drug we found works on mitochondria. In addition, among the drugs that have already been used for other purposes, we are evaluating those that improve the mitochondrial functions of Parkinson's disease patients by combining iPS cells from Parkinson's disease patients with a fly model of Parkinson's disease (Yamaguchi, Stem Cell Rep 2020).
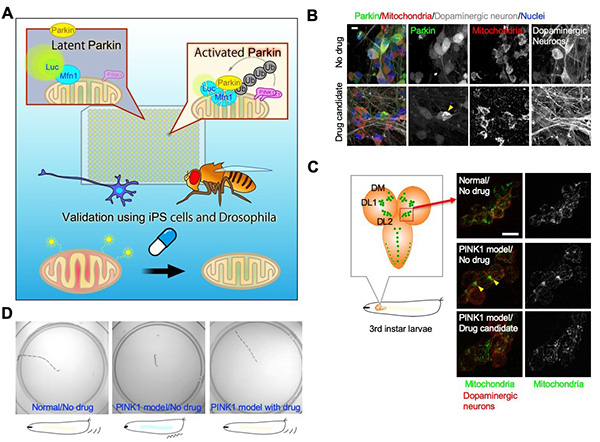
Figure 22. Searching for drugs that activate
Parkin.
(A) We developed a cell-based screening system to detect the activity of Parkin, a ubiquitin
ligase. 45,000 small molecule compounds were screened and two candidates were identified. (B) In
dopaminergic neurons generated from human iPS cells, Parkin, which is localized in the
cytoplasm, was found to translocate to the mitochondria upon drug treatment (yellow arrowheads).
This indicates that Parkin is activated. Scale: 10 μm. (C, left) Schematic representation of
dopaminergic neurons (green) in the brain (orange) of Drosophila third instar larvae.
(C, right) Photograph of dopaminergic neurons (red) in the DL2 nucleus. In a Parkinson's disease
model with impaired PINK1 function (PINK1 model fly), Parkin does not work fully and
mitochondria aggregation is observed (yellow arrowheads). Drug administration restores normal
mitochondrial morphology. Scale: 10 μm. (D) PINK1 model fly larvae have poor movement due to
impaired mitochondrial function. After administration of the drug, the movement improves. The
photos show the traces of larval movement for 2 minutes after being placed in the center of
petri dishes.
How a high-fat diet puts you at risk for Alzheimer's disease
Alzheimer's disease is a neurodegenerative disease caused
by the accumulation of
ß-amyloid and aggregated tau fibrils in the hippocampus, which stores short-term memory. The
molecular relationship between
ß-amyloid formation and tau fibrillization is unclear although tau fibrillization is known to
occur after ß-amyloid accumulation.
Like α-synuclein aggregation in Parkinson's disease, aggregated tau has been observed to spread
throughout the brain like a prion protein.
Epidemiological studies have shown that diabetes is a risk factor for Alzheimer's disease.
However, the reason for this was unknown. Mice on
a high-fat diet develop diabetes, and the genes whose expression is altered during this process
were investigated (Elahi, Hum Mol Genet. 2021).
SGK1 activates the tau kinase GSK3β, which promotes tau aggregation, whereby the learning and
memory abilities of mice fed a high-fat diet decreased (Fig. 23).
When the mice were treated with an inhibitor of SGK1, their learning and memory abilities were
restored. This study suggests that SGK1 inhibitors may be
a potential drug for Alzheimer's disease (This study was conducted in collaboration with the
Department of Diagnosis, Prevention and Treatment of Dementia,
Juntendo University Graduate School of Medicine)
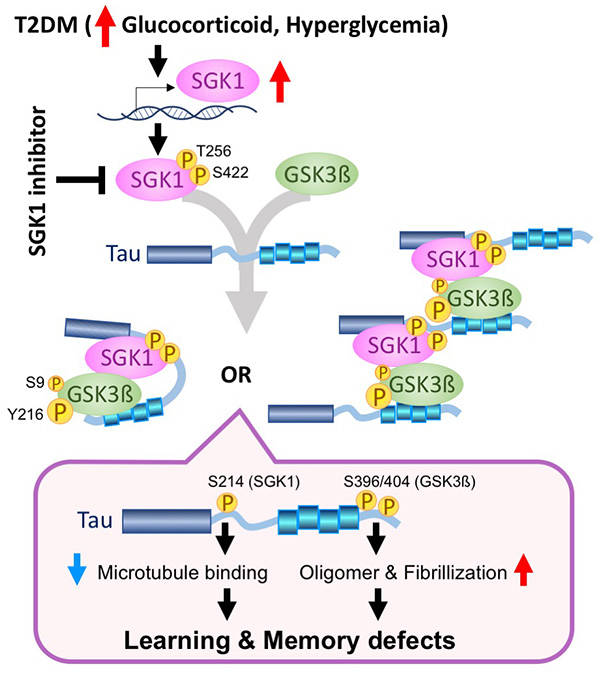
Figure 23. Mechanism by which tau aggregation, a
cause of Alzheimer's disease, progresses in diabetes.
When mice are continuously fed a high-fat diet, stress-response hormones
(glucocorticoids) and blood glucose in the blood rise, producing insulin resistance
(inability to respond to insulin), which is the cause of diabetes. Glucocorticoids and
hyperglycemia
upregulate the expression of the protein kinase SGK1. The increased expression of SGK1 activates
the
tau kinase GSK3β and phosphorylates tau at Ser214. GSK3β, activated by SGK1, phosphorylates tau
at Ser396/404
and causes tau aggregation and fibrillization. An SGK1 inhibitor reduces the decline in learning
memory in mice
fed a high-fat diet (Figure adapted from Elahi, Hum Mol Genet. 2021).
Parkinson's disease-related enzymes that monitor mitochondria
Parkin and PINK1 are both causative genes for juvenile
Parkinson's disease (
Fig. 2-4
 ×
and
Table 1
×
). When these genes fail to function, Parkinson's disease develops at the age of 10-40
years. Mitochondria-resident protein PINK1 is a protein kinase for Parkin and ubiquitin. Parkin
is a ubiquitin ligase that is involved in removal of damaged mitochondria. Mutations in the
Parkin or PINK1 genes are thought to cause neuronal death of midbrain dopaminergic neurons by
failure of the monitoring of damaged mitochondria. However, it is not yet known why the absence
of the Parkin or PINK1 genes causes neurodegeneration at a young age.
×
and
Table 1
×
). When these genes fail to function, Parkinson's disease develops at the age of 10-40
years. Mitochondria-resident protein PINK1 is a protein kinase for Parkin and ubiquitin. Parkin
is a ubiquitin ligase that is involved in removal of damaged mitochondria. Mutations in the
Parkin or PINK1 genes are thought to cause neuronal death of midbrain dopaminergic neurons by
failure of the monitoring of damaged mitochondria. However, it is not yet known why the absence
of the Parkin or PINK1 genes causes neurodegeneration at a young age.
We have reported that Parkin is activated by the phosphorylation by PINK1 (Shiba-Fukushima,
PLoS Genet. 2014a, Shiba-Fukushima, PLoS
Genet. 2014b). We found that Parkin attaches a small protein called
ubiquitin to itself during the activation (so-called self-ubiquitination). When we introduced
self-ubiquitination-resistant mutations to Parkin in Drosophila, ubiquitin ligase
activity of Parkin was reduced (Fig. 24A ). Detailed examination revealed that the ubiquitin
attached to Parkin is phosphorylated by PINK1, which promotes Parkin activation (Fig. 24B). The
ubiquitination of lysine 27 is suggested to be important for Parkin function because Parkinson's
disease patients with Parkin K27N exist (Liu, Hum Mol Genet. 2022).
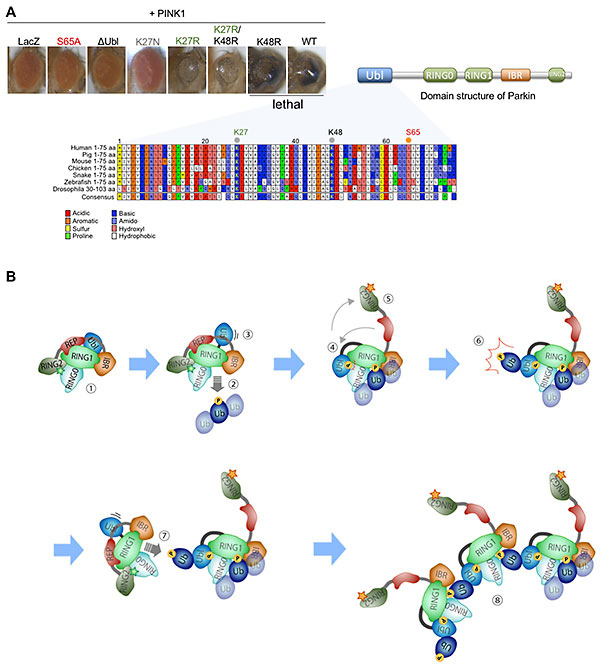
Figure 24. Ubiquitination of Parkin at lysine 27
promotes Parkin's ubiquitin ligase activity
(A) Parkin has an N-terminal ubiquitin-like domain (Ubl). Phosphorylation of serine 65 (S65) in
the Ubl by PINK1 activates Parkin and promotes mitochondrial degradation. Overexpression of
normal (wild-type, WT) Parkin together with PINK1 led to mitochondrial degradation in the eyes,
resulting in pupal lethality. On the other hand, when Parkin with S65 replaced by alanine (S65A,
phospho-resistant mutant), Parkin lacking Ubl (ΔUbl), or a mock protein (LacZ) was expressed
together with PINK1, the eyes develop normally. When lysine 27 (K27) and/or K48, were replaced
with arginine (R) not to self-ubiquitinate, flies expressing K27R Parkin, but not K48R Parkin,
survived under the same condition. This means that K27R Parkin is less active in degrading
mitochondria. Parkinson’s disease-linked K27N mutant also resulted in normal fly eyes. (B) A
model of Parkin activation via K27 ubiquitination. ① Parkin is inactive at steady state. ② When
mitochondria are damaged, activated PINK1 phosphorylates ubiquitin (Ub) chains on the
mitochondria (P). Parkin binds to the phosphorylated ubiquitin chains, and conformational change
of Parkin occurs (③-⑤). ⑥ The conformational change activates ubiquitin ligase, which adds
ubiquitin to K27 in its own Ubl. PINK1 phosphorylates ubiquitin at K27 (P). ⑦ Nearby Parkin in
the inactive state binds to the phospho-ubiquitin at K27 and is activated. ⑧ Steps ⑥-⑦ are
repeated to form a Parkin activation complex.
Mutations in α-Synuclein and lipids at risk for Parkinson's disease
The aggregation of α-Synuclein is believed to be the most
common cause of Parkinson's disease (see also Is Parkinson's disease a prion
disease?). α-Synuclein is thought to bind to and to be stabilized (assume a certain
shape) on the phospholipid membranes of synaptic vesicles. We found two Japanese Parkinson's
disease families carrying α-Synuclein V15A mutation in collaboration with the Department of
Neurology, Nagoya University Graduate School of Medicine (Daida, Mov
Disord. 2022). α-Synuclein V15A had a weaker binding property to
phospholipid vesicles mimicking synaptic vesicles and tends to aggregate when detached from
phospholipid vesicles.
The above observation suggests that α-Synuclein becomes a risk of Parkinson's disease when
α-Synuclein has a reduced binding activity to the phospholipid membranes of synaptic vesicles
(Fig. 25). Possible factors contributing to the reduced binding to phospholipids include
mutations in α-Synuclein itself and changes in the lipid composition of synaptic vesicles (see
also
Fig. 17
×
). The lipid composition in synaptic vesicles may also be affected by daily
diet. Research is currently underway to determine which lipids may reduce (or increase) the risk
of developing Parkinson's disease. Our goal is to realize that "When you continue to take
certain lipids, you will reduce your risk of developing Parkinson's disease."
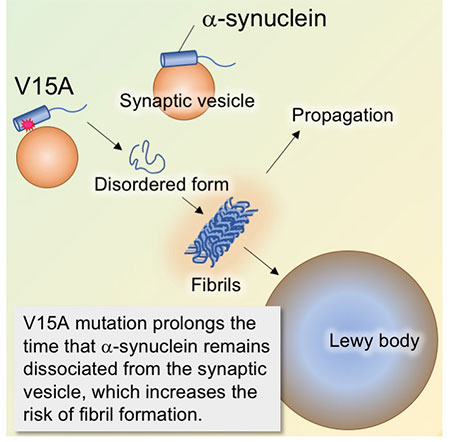
Figure 25. Molecular mechanism by which
α-Synuclein V15A mutations is a risk for Parkinson's disease.
V15A mutation weakens the binding of α-Synuclein to synaptic vesicles. Free α-Synuclein in the
cytoplasm does not assume a constant form, increasing the risk of fibril formation. The large
aggregation of α-Synuclein fibrils is thought to be the main component of Lewy bodies seen in
Parkinson's disease.
LRRK2 mutations at risk for Parkinson's disease and its relationship to α-Synuclein
LRRK2, together with α-Synuclein, is an important risk gene for Parkinson's disease. LRRK2 is a
protein kinase with multiple domains (Fig. 26A, see also
Fig. 9
×
). Several pathogenic mutations in these domains have been found worldwide,
and most of pathogenic mutations increase its kinase activity. The brain pathology of LRRK2
mutations is characterized by heterogeneous pathology, including the presence or absence of Lewy
bodies, which are aggregates of α-Synuclein, and the accumulation of phosphorylated tau, which
is characteristic of Alzheimer's disease.
The G2385R variant (a missense mutation in which the glycine residue at position 2385 is
replaced by an arginine residue) in the C-terminal WD40 domain of LRRK2 is also present in
healthy individuals, but is reported to double the risk of Parkinson's disease in Asian people.
We firstly reported the pathological and biochemical analyses of a patient with the G2385R
mutation (Tezuka, NPJ Parkinsons Dis 2022). The G2385R variant was
associated with elevated kinase activity. The accumulation of both Lewy bodies and
phosphorylated tau was observed in the brain autopsy (Fig. 26B). On the other hand, there was no
correlation between the brain regions with high LRRK2 kinase activity and the brain regions with
prominent Lewy body accumulation (Fig. 26C). LRRK2 is also thought to be involved in brain
inflammation. However, the activation of astrocytes and microglia, signs of inflammation, was
moderate (Fig. 26B). Our results suggest that elevated LRRK2 kinase activity may have a role in
promoting brain aging rather than being directly involved in α-synuclein aggregation and
propagation.
Since elevated LRRK2 kinase activity leads to dopaminergic neurodegeneration, the cause of
Parkinson's disease, LRRK2 inhibitors have been developed worldwide. However, LRRK2 is also
known to play important roles in the lungs and kidneys, and complete inhibition of LRRK2 is
expected to affect the functions of lungs and kidneys. Thus, the elucidation of the
patho/physiological roles of LRRK2 in the brain and the moderate regulation of LRRK2 enzyme
activity would be the challenges in overcoming Parkinson's disease caused by LRRK2 mutations.
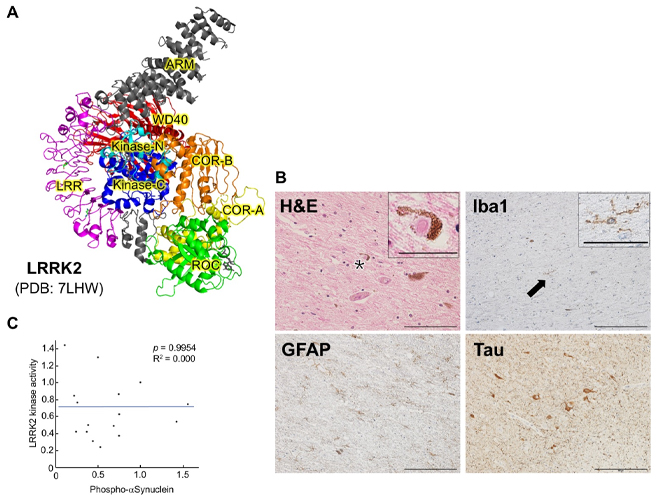
Figure 26. The first report of brain pathology for
LRRK2 G2385R variant.
(A) 3D structure of LRRK2 deduced by cryo-EM (drawn from data in the Protein Data Bank![]() ). The WD40 spatially surrounds
N-terminal lobe of the kinase domain (Kinase-N). G2385R is expected to destabilize the WD40
structure and affect the kinase domain. (B) A case with LRRK2 G2385R showed typical Lewy body
pathology. Hematoxylin-eosin (H&E) staining reveals typical Lewy bodies (Inset is a magnified
image of a region containing an asterisk). Microglia (Inset is a magnified image of a region
containing an arrow) and astrocytes were stained with antibodies against Iba1 and GFAP,
respectively. Phosphorylated tau (Tau) is also markedly accumulated. Scale bars: H&E staining,
100 µm; Iba1, GFAP, Tau, 200 µm; magnified images, 25 µm. (C) No correlation between LRRK2
kinase activity and α-Synuclein aggregation. The horizontal axis of the graph shows
quantification of phosphorylated α-Synuclein in each brain region; the vertical axis is the
quantification of phosphorylated Rab10 in the corresponding brain region. Phosphorylated
α-Synuclein and phosphorylated Rab10 are used as indicators of α-Synuclein aggregation (Lewy
bodies) and LRRK2 kinase activity, respectively.
). The WD40 spatially surrounds
N-terminal lobe of the kinase domain (Kinase-N). G2385R is expected to destabilize the WD40
structure and affect the kinase domain. (B) A case with LRRK2 G2385R showed typical Lewy body
pathology. Hematoxylin-eosin (H&E) staining reveals typical Lewy bodies (Inset is a magnified
image of a region containing an asterisk). Microglia (Inset is a magnified image of a region
containing an arrow) and astrocytes were stained with antibodies against Iba1 and GFAP,
respectively. Phosphorylated tau (Tau) is also markedly accumulated. Scale bars: H&E staining,
100 µm; Iba1, GFAP, Tau, 200 µm; magnified images, 25 µm. (C) No correlation between LRRK2
kinase activity and α-Synuclein aggregation. The horizontal axis of the graph shows
quantification of phosphorylated α-Synuclein in each brain region; the vertical axis is the
quantification of phosphorylated Rab10 in the corresponding brain region. Phosphorylated
α-Synuclein and phosphorylated Rab10 are used as indicators of α-Synuclein aggregation (Lewy
bodies) and LRRK2 kinase activity, respectively.
The relationship between the Parkinson's disease-causative gene CHCHD2 and amyotrophic lateral sclerosis (ALS)
CHCHD2, which has been identified as a gene that causes Parkinson's disease, stabilizes cytochrome c in the mitochondrial respiratory chain complex and regulates electron transport (see also the Section on "Regulation of mitochondrial function by Parkinson's disease-causing genes").
On the other hand, CHCHD10 has been reported as a causative gene for ALS and frontotemporal dementia (FTD). CHCHD10 and CHCHD2 are related to each other like twins. In other words, it is thought that they diverged into two copies from one gene in the
evolutionary process, and the amino acid sequences of both molecules are similar. We also examined the genomic DNA of patients with ALS, considering the possibility that mutations in CHCHD2 could also be a risk factor for ALS (Ikeda, PNAS Nexus 2024). As a result, we found a patient with an amino acid substitution of proline to leucine at position 14 (P14L). In typical ALS, the RNA-binding protein TDP-43 aggregates and accumulates in motor neurons. The patient with CHCHD2 P14L also had TDP-43
aggregation. CHCHD2 is present in the mitochondrial intermembrane space and forms a complex with the twin molecule CHCHD10. However, the P14L mutation weakened the binding to CHCHD10, making CHCHD2 more likely to leak from the mitochondria into the cytoplasm.
As a result, the mitochondrial Ca2+ buffering function was reduced, and the cleavage and aggregation of TDP-43 was accelerated (for details, please refer to Figure 27 and Ikeda, PNAS Nexus 2024).
In this study, we observed an increased expression of CHCHD2 and CHCHD10 mRNA in patients with sporadic (without a family history) ALS. Since CHCHD2 and CHCHD10 expression increases when mitochondria are stressed, this observation suggests
that mitochondrial damage also occurs in patients with sporadic ALS. There is a disease called Kii Amyotrophic Lateral Sclerosis/Parkinsonism-Dementia Complex (Kii ALS/PDC). It is a local disease that is often seen in the Kii Peninsula in Japan, but the
genetic and environmental factors remain unclear. A recent study reports that astrocytes differentiated from iPS cells of patients with Kii ALS/PDC show decreased expression of CHCHD2 and mitochondrial dysfunction (PMID: 38750212). As such, CHCHD2 is
suggested to be involved in the pathogenesis of both Parkinson's disease and ALS.
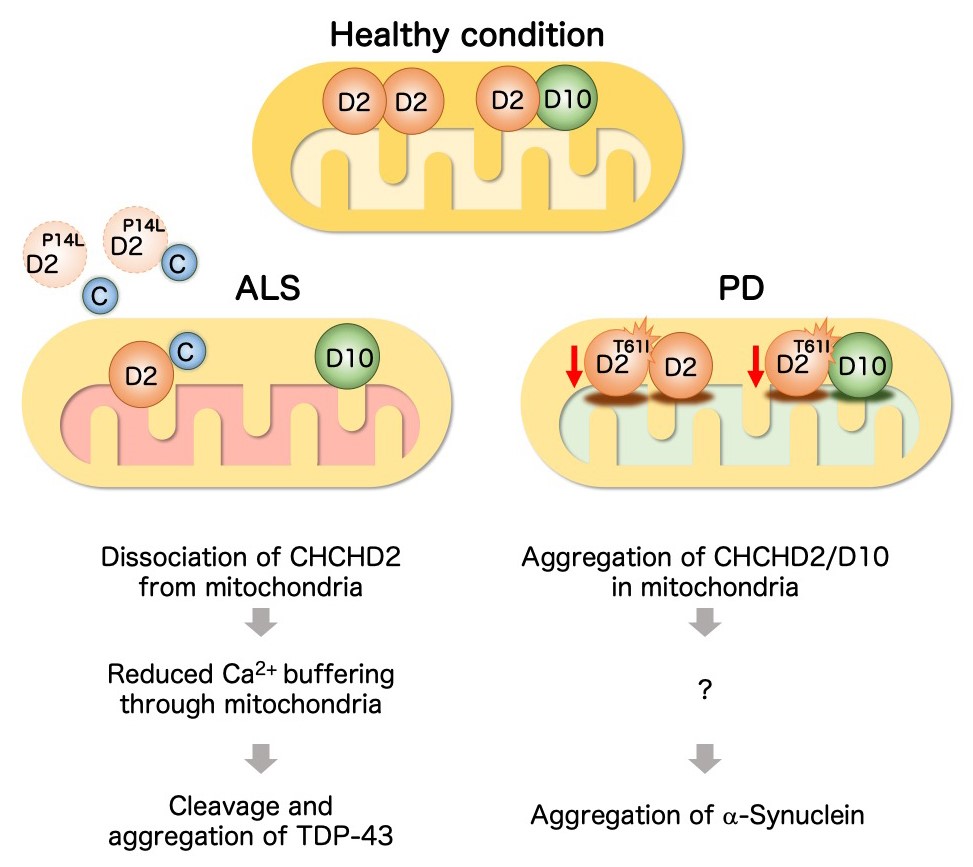
Figure 27. Molecular mechanism by which CHCHD2 mutations cause ALS and Parkinson's disease (Working hypothesis)
(Left) In motor neurons, cytoplasmic Ca2+ concentration increase in response to neuronal activity, but if the Ca2+ concentration remains high for a sustained period of time, this situation can lead to neurotoxicity. The endoplasmic
reticulum and mitochondria have a buffering function that absorbs the increased cytoplasmic Ca2+. The P14L mutation detected in ALS promotes the release of CHCHD2 from mitochondria into the cytoplasm. As a result, the mitochondrial Ca2+ buffering capacity is reduced. If the cytoplasmic Ca2+ level remains high for a sustained period of time, CHCHD2 and cytochrome C (C) are further released from the mitochondria. The free cytochrome C activates caspases. In addition, high concentrations
of cytoplasmic Ca2+ activate calpain. Caspases and calpain cleave TDP-43, and promote aggregation of its C-terminal intrinsically disordered region. (Right) The T61I mutation seen in Parkinson's disease causes insolubilization of CHCHD2 and
its binding partner CHCHD10. The insolubilized CHCHD2-CHCHD10 causes mitochondrial stress, which promotes α-Synuclein aggregation.