丂丂僞僀僩儖傪僋儕僢僋傪偟傑偡偲撪梕偑昞帵偝傟傑偡
堚揱惈僷乕僉儞僜儞昦
|
丂僷乕僉儞僜儞昦偺5-10%偵堚揱惈偺傕偺偑尒傜傟傑偡丅嵟嬤偦偺尨場堚揱巕孮偑柧傜偐偵偝傟偮偮偁傝丄暘巕惗暔妛揑傾僾儘乕僠偑壜擻偲側傝傑偟偨乮昞1乯丅偙傟傜堚揱巕偺曄堎偵傛傝拞擼崟幙偺僪乕僷儈儞恄宱偑曄惈偟丄僷乕僉儞僜儞昦偑敪徢偡傞偙偲偑暘偐偭偰偄傑偡偑丄偦傟偧傟偺堚揱巕嶻暔偺曄堎偑恄宱曄惈偵偳偺傛偆偵婑梌偟偰偄傞偺偐丄嫟捠偺恄宱曄惈儊僇僯僘儉偑懚嵼偡傞偺偐偼傛偔暘偐偭偰偄傑偣傫丅
恾1. PINK1,
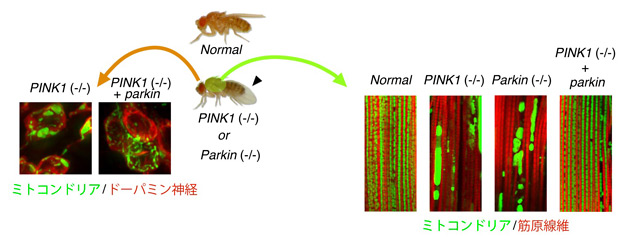
Parkin儌僨儖僔儑僂僕儑僂僶僄偼儈僩僐儞僪儕傾曄惈傪帵偡
僔儑僂僕儑僂僶僄傕PINK1丄Parkin堚揱巕傪帩偪丄偙傟傜偺僲僢僋傾僂僩乮-/-乯僔儑僂僕儑僂僶僄偼丄嬝擏乮塃乯傗僪乕僷儈儞恄宱乮嵍乯偺儈僩僐儞僪儕傾乮幨恀撪丄椢怓乯偑曄惈乮嫄戝側儈僩僐儞僪儕傾偺夠偑曄惈憸乯偟丄塇偑壓悅偟傑偡乮栴摢乯乮Imai,
PLoS Genet.
2010乯丅PINK1僲僢僋傾僂僩僔儑僂僕儑僂僶僄偵丄奜偐傜Parkin堚揱巕傪擖傟傞偲丄儈僩僐儞僪儕傾偺曄惈偑夵慞偟傑偡乮偦傟偧傟偺幨恀偺塃抂乯丅
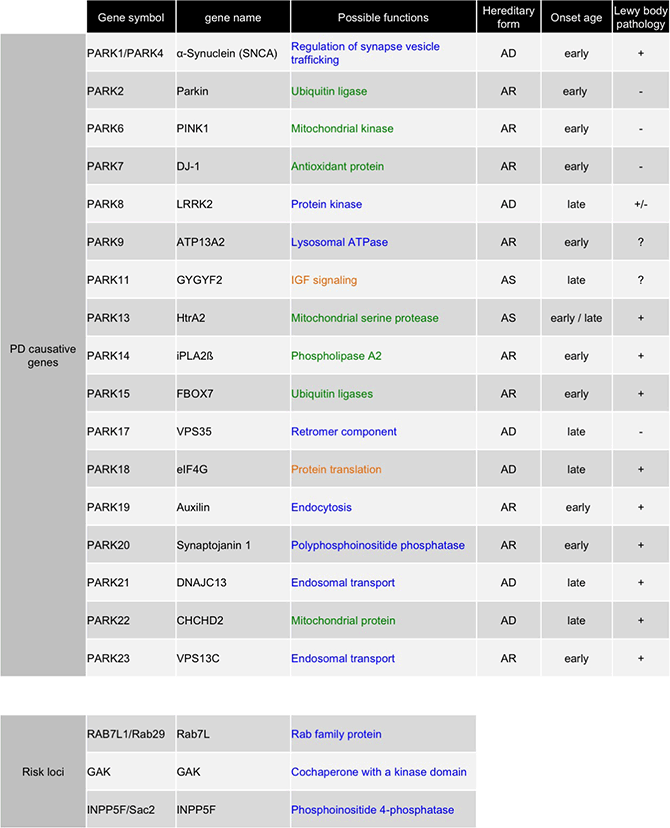
昞1.
僷乕僉儞僜儞昦尨場堚揱巕
AD; 忢愼怓懱桪惈堚揱丄AR; 忢愼怓懱楎惈堚揱丄AS;
姶庴惈堚揱巕丅丠偼枹摨掕丒枹夝柧傪堄枴偟傑偡丅PARK21偼丄摨堦偺壠宯傛傝丄2偮偺堚揱巕(DNAJC13丄TMEM230)偺僶儕傾儞僩偑尒偮偐偭偰偍傝丄偳偪傜偑昦場堚揱巕偱偁傞偐偼晄柧偱偡丅惵偼彫朎桝憲偵娭楢偡傞堚揱巕丄椢偼儈僩僐儞僪儕傾偵娭楢偺偁傞堚揱巕丄僆儗儞僕偼偦偺懠偺婡擻傪傕偮堚揱巕偱偡丅堚揱巕嶻暔偼條乆側婡擻傪傕偮僞儞僷僋幙偱偡偑丄偙傟傜偑撈棫偵婡擻偟偰偄傞偺偐丄偁傞偄偼嫟捠偺昦棟宱楬偱嶌梡偟偰偄傞偺偐丄偲偄偆偙偲偼夝柧偡傋偒廳梫側壽戣偱偡丅変乆偼僔儑僂僕儑僂僶僄傪儌僨儖偲偟偰丄Parkin偲PINK1偵堚揱揑憡屳嶌梡偑偁傞偙偲傪柧傜偐偵偟傑偟偨乮Yang,
PNAS 2006乯丅
|
僷乕僉儞僜儞昦儌僨儖僔儑僂僕儑僂僶僄
| |
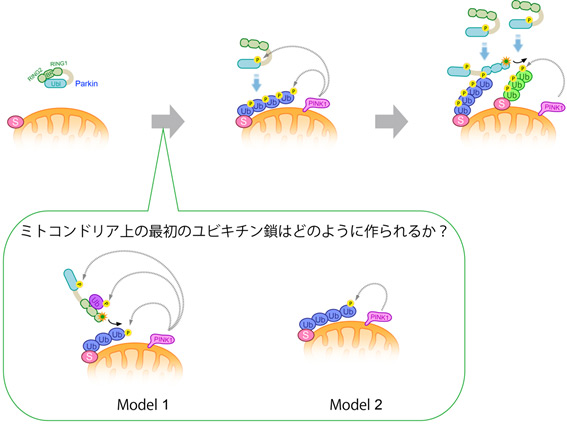
丂偙偺栤戣傪柧傜偐偵偡傞偨傔丄暘巕堚揱妛揑夝愅偵揔偟庻柦偑抁偄僔儑僂僕儑僂僶僄傪梡偄偰夝愅傪峴偭偰偄傑偡乮恾1, 5, 6乯丅僔儑僂僕儑僂僶僄傕僪乕僷儈儞恄宱傪帩偪丄僸僩偲摨偠傛偆偵榁壔偟傑偡丅椺偊偽丄塣摦擻椡丒擣抦擻椡偺掅壓丄帀幙偺巁壔丄惗怋擻椡偺掅壓側偳偑尒傜傟傑偡丅儅僂僗傪儌僨儖偲偟暋悢偺僷乕僉儞僜儞昦堚揱巕偺娭學傪柧傜偐偵偡傞偵偼悢擭偐偐傝傑偡偑丄僔儑僂僕儑僂僶僄傪梡偄傞偲侾擭埲撪偵偦偺娭學傪抦傞偙偲偑偱偒傑偡丅
丂巹偨偪偼丄儌僨儖僴僄傪巊偭偰丄昞侾偵偁傞儈僩僐儞僪儕傾偵娭學偡傞僷乕僉儞僜儞昦尨場堚揱巕乮恾7乯偲彫朎桝憲偵娭學偡傞僷乕僉儞僜儞昦堚揱巕乮恾8-12乯偺婡擻偲偦偺曄堎偵傛傞恄宱曄惈偺儊僇僯僘儉傪柧傜偐偵偟傛偆偲庢傝慻傫偱偄傑偡丅
|
| |
|
僾儘僥僆儈僋僗夝愅
| |
丂暘巕堚揱妛揑夝愅偺傒偱偼丄僷乕僉儞僜儞昦尨場堚揱巕偐傜嶌傜傟傞僞儞僷僋幙偺摥偒偼暘偐傝傑偣傫丅僷乕僉儞僜儞昦偺昦場曄堎偑僞儞僷僋幙偺摥偒偵偳偺傛偆偵嶌梡偡傞偐偼丄姅壔嵶朎丄弶戙攟梴恄宱偺攟梴傗僾儘僥僆儈僋僗夝愅偵傛傝尋媶偟偰偄傑偡丅傑偨丄僷乕僉儞僜儞昦儌僨儖儅僂僗傪巊偭偰丄僞儞僷僋幙偺摥偒傪夝愅偡傞偙偲傕偁傝傑偡丅儈僩僐儞僪儕傾偺婡擻堐帩偵摥偔僷乕僉儞僜儞昦尨場堚揱巕PINK1偲Parkin偺栶妱偼丄攟梴嵶朎偱傕柧傜偐偵偟偰偒傑偟偨乮恾2-4乯乮Shiba-Fukushima,
Sci Rep. 2012;
PLoS Genet. 2014b乯丅
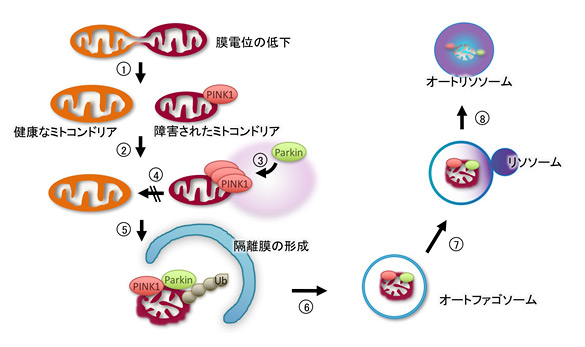
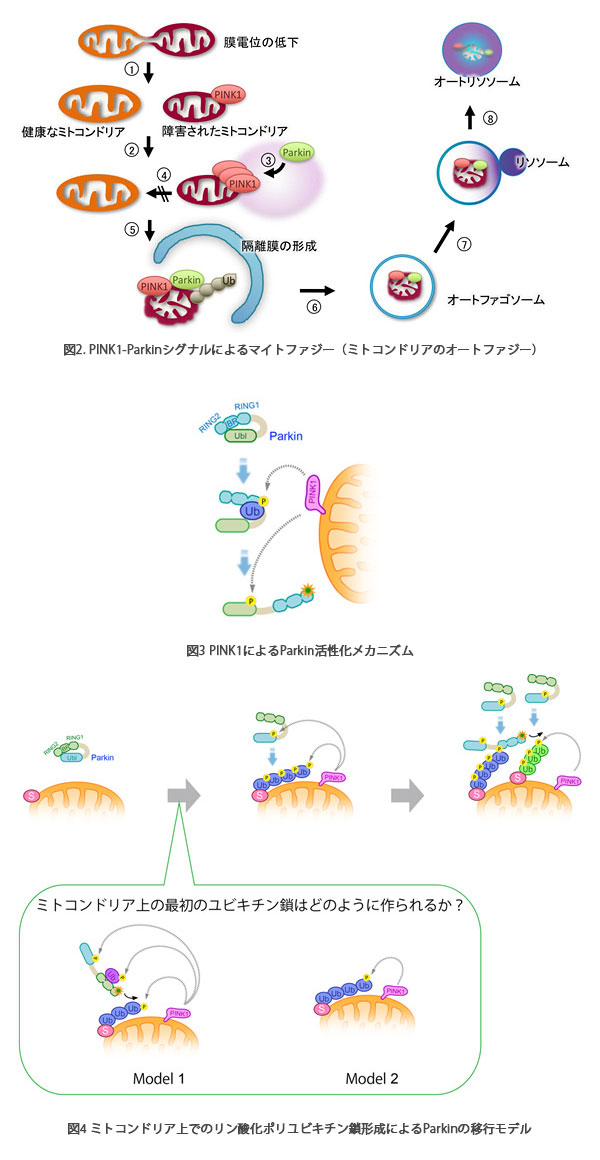
恾2.
PINK1-Parkin僔僌僫儖偵傛傞儅僀僩僼傽僕乕乮儈僩僐儞僪儕傾偺僆乕僩僼傽僕乕乯
PINK1-Parkin僔僌僫儖偼晄椙儈僩僐儞僪儕傾傪埲壓偺傛偆側僗僥僢僾傪宱偰彍嫀偡傞丅
嘆枌揹埵偺掅壓偟偨晄椙儈僩僐儞僪儕傾偼暘抐偝傟丄嘋嵞梈崌偟側偄丅嘇枌揹埵偺掅壓偟偨儈僩僐儞僪儕傾忋偱PINK1偑拁愊偟丄僉僫乕僛妶惈偑妶惈壔偡傞丅嘍PINK1偵傛傝Parkin偑妶惈壔偝傟丄Parkin偼嵶朎幙偐傜儈僩僐儞僪儕傾傊堏峴偡傞丅師偵Parkin偼儈僩僐儞僪儕傾奜枌僞儞僷僋幙孮傪儐價僉僠儞(Ub)壔偡傞丅嘐乣嘒儐價僉僠儞壔傪庴偗偨儈僩僐儞僪儕傾偵TBK1,
Optineurin,
LC3側偳偺僆乕僩僼傽僕乕娭楢僞儞僷僋幙偑儕僋儖乕僩偝傟丄晄椙儈僩僐儞僪儕傾偼僆乕僩僼傽僕乕宱楬偱暘夝偝傟傞丅
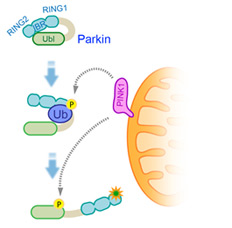
恾3
PINK1偵傛傞Parkin妶惈壔儊僇僯僘儉
掕忢帪丄Parkin偼嵶朎幙偱晄妶惈側忬懺偲偟偰僐儞僷僋僩偵愜傝忯傑傟偰偄傞丅儈僩僐儞僪儕傾婡擻忈奞偵傛傝枌揹埵偑掅壓偡傞偲PINK1偑妶惈壔偡傞丅妶惈壔PINK1偵傛傝儕儞巁壔乮Ⓟ乯偝傟偨儐價僉僠儞(Ub)偑Parkin偺RING1-IBR椞堟偵擖傝崬傓偙偲偵傛傝Parkin偺峔憿偑娚傒丄師偵PINK1偑Parkin偺儐價僉僠儞條椞堟(Ubl)傪儕儞巁壔偡傞丅偙傟偵傛傝儐價僉僠儞儕僈乕僛妶惈拞怱偑業弌偟丄Parkin偑妶惈宆儐價僉僠儞儕僈乕僛偲側傞丅
恾4
儈僩僐儞僪儕傾忋偱偺儕儞巁壔億儕儐價僉僠儞嵔宍惉偵傛傞Parkin偺堏峴儌僨儖
Parkin偼晄妶惈宆偺儐價僉僠儞儕僈乕僛偲偟偰嵶朎幙偵嬊嵼偡傞乮嵍乯丅
儈僩僐儞僪儕傾偑懝彎偟枌揹埵偑掅壓偡傞偲丄PINK1偑拁愊丒妶惈壔偟丄Parkin偺儐價僉僠儞條僪儊僀儞傪儕儞巁壔偡傞丅儈僩僐儞僪儕傾忋偵億儕儐價僉僠儞嵔偑宍惉偝傟丄偝傜偵PINK1偑偙傟傪儕儞巁壔偡傞丅Parkin偼儕儞巁壔億儕儐價僉僠儞嵔偵恊榓惈偑偁傝丄儈僩僐儞僪儕傾傊嬊嵼壔偡傞乮拞墰乯丅
儕儞巁壔億儕儐價僉僠儞嵔偵寢崌偟偨Parkin偼丄儐價僉僠儞儕僈乕僛妶惈偑妶惈壔偟丄儈僩僐儞僪儕傾奜枌僞儞僷僋幙偵億儕儐價僉僠儞嵔傪宷偖丅偝傜偵丄偙偺億儕儐價僉僠儞嵔傪PINK1偑儕儞巁壔偡傞偙偲偵傛傝丄儈僩僐儞僪儕傾奜枌忋偱儕儞巁壔億儕儐價僉僠儞嵔偺憹暆斀墳偑婲偙傝丄Parkin偺恦懍側儈僩僐儞僪儕傾堏峴偲妶惈壔偑払惉偝傟傞乮塃乯丅
Ub;儐價僉僠儞, P;儕儞巁壔, S;
儈僩僐儞僪儕傾奜枌忋偺儐價僉僠儞壔婎幙僞儞僷僋幙丅
(壓偺埻傒)嵟弶偺儕儞巁壔億儕儐價僉僠儞嵔偼偳偺傛偆偵宍惉偝傟傞偐丠
PINK1偵傛傝儕儞巁壔傪庴偗偨儌僲儐價僉僠儞偵傛偭偰Parkin偑妶惈壔偝傟傞丅妶惈壔偝傟偨Parkin偼帺桼奼嶶偵傛傝儈僩僐儞僪儕傾忋偺婎幙傪儐價僉僠儞壔偟丄偦傟傪PINK1偑儕儞巁壔廋忺偡傞壜擻惈乮儌僨儖侾乯偲丄Parkin埲奜偺儐價僉僠儞儕僈乕僛偵傛偭偰惗棟揑偵儐價僉僠儞壔廋忺傪庴偗傞儈僩僐儞僪儕傾奜枌僞儞僷僋幙偺儐價僉僠儞嵔偑PINK1偵傛傝儕儞巁壔偝傟丄偦傟偑Parkin偺儈僩僐儞僪儕傾嬊嵼偺嵟弶偺懌応偲偟偰巊傢傟傞壜擻惈乮儌僨儖俀乯偑峫偊傜傟傞乮Shiba-Fukushima,PLoS
Genet. 2014b; 恾偼丄崱嫃傜
幚尡堛妛 2014傛傝揮嵹乯丅
|
椪彴僒儞僾儖偱偺尋媶
| |
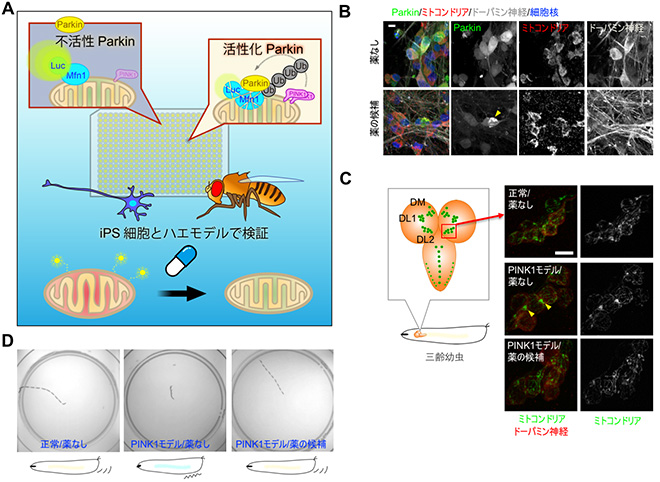
弴揤摪戝妛恄宱妛島嵗偲嫤椡偟偰丄僷乕僉儞僜儞昦姵幰偝傫偐傜扨棧偟偨攟梴嵶朎乮iPS嵶朎乯傕梡偄偨傝傕偟傑偡丅僔儑僂僕儑僂僶僄傗僾儘僥僆儈僋僗夝愅偱暘偐偭偨偙偲偑丄姵幰偝傫偺懱偺拞偱婲偙偭偰偄傞偐偳偆偐妋擣偡傞偨傔偱偡丅椺偊偽丄僔儑僂僕儑僂僶僄儌僨儖偱尒偮偗偨Parkin偺儐價僉僠儞壔婎幙偵Miro偑偁傝傑偡丅Miro偼儈僩僐儞僪儕傾偺桝憲偵昁梫側僞儞僷僋幙偱丄Parkin偑妶惈壔偝傟傞偲儈僩僐儞僪儕傾偺恄宱嵶朎偱偺桝憲偑巭傑傝傑偡丅僴僄偱娤嶡偟偨帠徾偑丄僸僩偺iPS嵶朎偐傜嶌偭偨僪乕僷儈儞恄宱偱傕嵞尰偱偒傑偟偨乮恾6乯丅偝傜偵丄Parkin偵曄堎偺偁傞姵幰偝傫偺僪乕僷儈儞恄宱偱偼晄椙儈僩僐儞僪儕傾偺桝憲偑巭傑傝偵偔偔側偭偰偄傑偟偨乮Shiba-Fukushima,
Hum Mol Genet. 2017乯丅
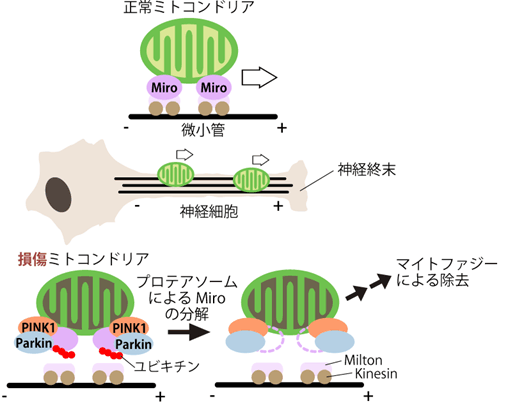
恾5
PINK1-Parkin偵傛傞Miro偺埨掕惈惂屼偑丄恄宱偺儈僩僐儞僪儕傾傪娗棟偡傞
Miro偼儈僩僐儞僪儕傾偺旝彫娗桝憲傪扴偄丄恄宱偱偼恄宱廔枛傑偱儈僩僐儞僪儕傾傪塣傇丅
懝彎偟偨儈僩僐儞僪儕傾偱偼PINK1偑Parkin傪妶惈壔偟丄Miro傪暘夝偡傞丅偙傟偵傛傝丄懝彎偟偨儈僩僐儞僪儕傾偺恄宱廔枛傊偺桝憲傪杊巭偡傞丅恄宱嵶朎懱偵棷傔傜傟偨儈僩僐儞僪儕傾偼儅僀僩僼傽僕乕偱暘夝偝傟傞偲峫偊傜傟傞乮Liu,PLoS
Genet. 2011乯丅
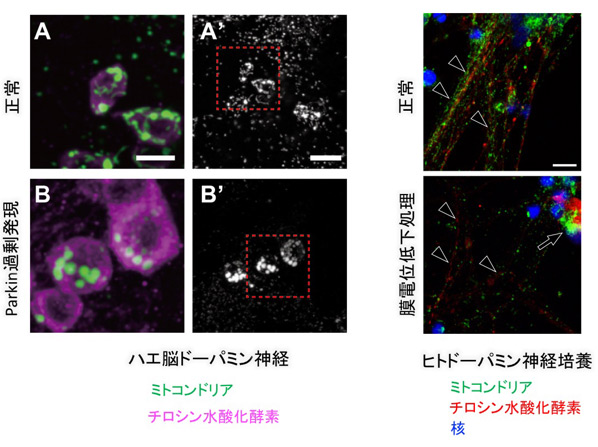
恾6
PINK1-Parkin偵傛傞僪乕僷儈儞恄宱嵶朎偱偺儈僩僐儞僪儕傾桝憲
(A-B’)僴僄偺擼偺僪乕僷儈儞恄宱乮僠儘僔儞悈巁壔峺慺梲惈偺嵶朎乯丅嵍偺僀儊乕僕偼塃偺愒榞偺奼戝恾丅乮A’,
B’乯僪乕僷儈儞恄宱偺儈僩僐儞僪儕傾僔僌僫儖丅乮A,
A’乯惓忢側僪乕僷儈儞恄宱偺儈僩僐儞僪儕傾憸丅乮B,
B’乯Parkin傪夁忚偵敪尰偡傞偲Miro偺暘夝偑恑傒丄恄宱廔枛偺儈僩僐儞僪儕傾僔僌僫儖偑徚幐偡傞丅堦曽丄恄宱嵶朎懱偵抐曅壔偟偨儈僩僐儞僪儕傾偑拁愊偡傞乮Shiba-Fukushima,PLoS
Genet.
2014b乯丅僸僩iPS嵶朎偐傜暘壔偝偣偨僪乕僷儈儞恄宱偵偍偄偰丄枌揹埵掅壓張棟傪峴偆偲丄僴僄摨條丄恄宱幉嶕乮栴摢乯偺儈僩僐儞僪儕傾偑徚幐偟丄恄宱嵶朎懱偵拁愊偡傞乮栴報乯丅僗働乕儖:
5 µm (A, B)丄10 µm (A’,
B’)丄10 µm乮僸僩恄宱乯丅
|
僷乕僉儞僜儞昦尨場堚揱巕偵傛傞儈僩僐儞僪儕傾婡擻偺惂屼
| |
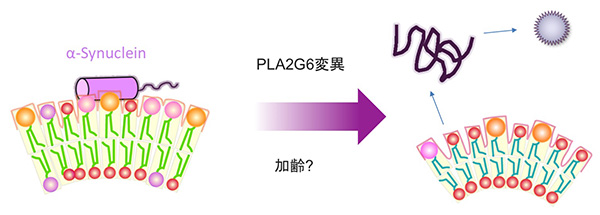
丂嵶朎彫婍姱偺堦偮偱偁傞儈僩僐儞僪儕傾偼丄惗懱偺僄僱儖僊乕偺尦偲側傞ATP偺崌惉丄帀幙戙幱丄揝戙幱丄嵶朎撪Ca2+擹搙偺挷愡丄嵶朎巰僔僌僫儖偺惂屼偲條乆側婡擻傪傕偪傑偡丅偙偺儈僩僐儞僪儕傾偺婡擻掅壓丄婡擻堎忢偑丄僷乕僉儞僜儞昦乮恾7丄昞1乯丄嬝堔弅惈懁嶕峝壔徢乮ALS乯丄慜摢懁摢宆擣抦徢乮FTD乯-ALS側偳偺恄宱曄惈幘姵偵娭梌偡傞偙偲偑丄堚揱妛揑側徹嫆偐傜柧傜偐偵側傝偮偮偁傝傑偡丅椺偊偽僷乕僉儞僜儞昦偵偍偄偰偼丄慜弎偺庒擭惈僷乕僉儞僜儞昦堚揱巕PINK1傗Parkin偑儈僩僐儞僪儕傾偺昳幙娗棟乮夡傟偨儈僩僐儞僪儕傾傪彍嫀偡傞偙偲乯偵娭傢偭偰偄傞偙偲偑帵偝傟偰偄傑偡丅堦曽丄斢敪惈僷乕僉儞僜儞昦堚揱巕CHCHD2偼丄儈僩僐儞僪儕傾屇媧嵔暋崌懱偺揹巕偺棳傟傪挷愡偟偰偄傑偡乮Meng,
Nat Commun.
2017乯丅CHCHD2偵僷乕僉儞僜儞昦曄堎偑擖傞偲丄揹巕偑楻傟偰巁壔僗僩儗僗偵偮偑傞偙偲偑暘偐偭偰偒傑偟偨丅
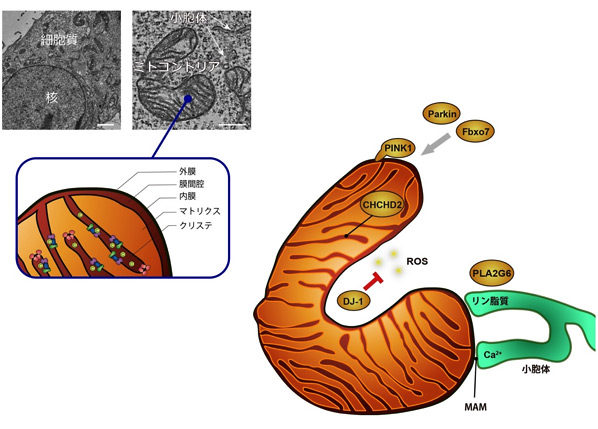
恾7
僷乕僉儞僜儞昦堚揱巕嶻暔偺儈僩僐儞僪儕傾偵偍偄偰憐掕偝傟偰偄傞婡擻恾
PINK1-Parkin偼儈僩僐儞僪儕傾偺昳幙娗棟偵娭傢傞丅Fbxo7傕Parkin偲嫟偵摥偔丅DJ-1偼丄儈僩僐儞僪儕傾偐傜敪惗偡傞妶惈巁慺庬乮ROS乯偺彍嫀偵娭傢傞丅CHCHD2偼儈僩僐儞僪儕傾枌娫峯偵懚嵼偟丄屇媧嵔暋崌懱偺妶惈堐帩偵娭梌偡傞丅PLA2G6偼儕儞帀幙偺儕儌僨儕儞僌傪夘偟偰丄儈僩僐儞僪儕傾偱敪惗偡傞ROS偵傛傝夁巁壔偝傟偨帀幙偺彍嫀丄彫朎懱偐傜棳擖偡傞Ca2+偺惂屼偵娭傢傞丅儈僩僐儞僪儕傾偼丄彫朎懱偲暔幙乮Ca2+,
帀幙側偳乯偺傗傝偲傝傪偟偰偍傝丄嬤愙晹埵乮30
nm埲壓乯偼偲偔偵Mitochondria Associated Membrane
(MAM)偲屇偽傟偰丄僞儞僷僋幙暋崌懱偱宷偑偭偰偄傞丅僸僩攟梴嵶朎偺揹巕尠旝嬀幨恀偺僗働乕儖:
2 兪m乮嵍乯丄500 nm (塃)丅乮恾偼丄崱嫃 擔杮椪喱 2017傛傝乯
|
僷乕僉儞僜儞昦尨場堚揱巕偵傛傞彫朎桝憲惂屼
| |
丂彫朎桝憲偲偼暵偠偨帀幙枌偱暔幙偑塣偽傟傞嵶朎撪偺尰徾偱偡丅嵶朎彫婍姱乮儈僩僐儞僪儕傾丄彫朎懱丄僑儖僕懱丄儕僜僜乕儉側偳乯娫偺暔幙偺傗傝庢傝偺偨傔偵巊傢傟傑偡丅傑偨嵶朎偺奜傊偺暔幙偺曻弌乮僄僉僜僒僀僩乕僔僗乯丄庢傝崬傒乮僄儞僪僒僀僩乕僔僗乯偵傕巊傢傟傑偡丅
丂僷乕僉儞僜儞昦尨場堚揱巕偺懡偔偑嵶朎撪彫朎偺桝憲偵娭傢偭偰偄傞偙偲偑柧傜偐偵側偭偰偒傑偟偨乮恾8, 9乯丅偙傟傜偺堚揱巕偼恄宱嵶朎丄旕恄宱嵶朎偄偢傟偱傕摥偄偰偄傞偙偲偑暘偐偭偰偄傑偡丅摿偵僷乕僉儞僜儞昦偺昦懺偵娭學偑怺偄僪乕僷儈儞恄宱偱偼丄慜僔僫僾僗偐傜偺僄儞僪僒僀僩乕僔僗乮恾10乯偵娭傢偭偰偄傞偙偲偑丄巹偨偪偍傛傃懠尋媶幰偺尋媶偐傜柧傜偐偵側傝偮偮偁傝傑偡乮Inoshita,
Hum Mol Genet. 2017,
Inoshita, J Genet.
2018乯丅
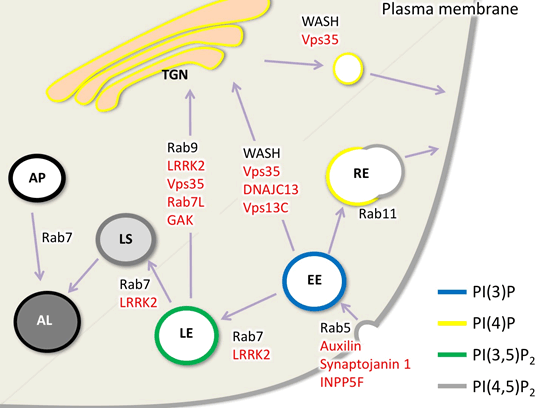
恾8
僷乕僉儞僜儞昦堚揱巕嶻暔偺彫朎桝憲偵偍偄偰憐掕偝傟偰偄傞婡擻恾
偙偙偱偼嵶朎枌偐傜偺僄儞僪僒僀僩乕僔僗丄僄儞僪僜乕儉丄儕僜僜乕儉丄僑儖僕懱娫偺彫朎桝憲傪帵偡丅愒帤偱帵偡傕偺偑僷乕僉儞僜儞昦尨場堚揱巕偁傞偄偼姶庴惈堚揱巕丅偦傟偧傟偺彫朎偼摿暿側儕儞巁壔廋忺傪庴偗偨僀僲僔僩乕儖儕儞帀幙偵傛傝埻傑傟偰偄傞丅TGN,
僩儔儞僗僑儖僕僱僢僩儚乕僋; AL, 僆乕僩儕僜僜乕儉; AP, 僆乕僩僼傽僑僜乕儉;
LS, 儕僜僜乕儉; EE, 弶婜僄儞僪僜乕儉; LE, 屻婜僄儞僪僜乕儉; RE,
儕僒僀僋儕儞僌僄儞僪僜乕儉丅乮恾偼丄Inoshita and Imai,
AIMS Mol Sci.
2015傛傝乯
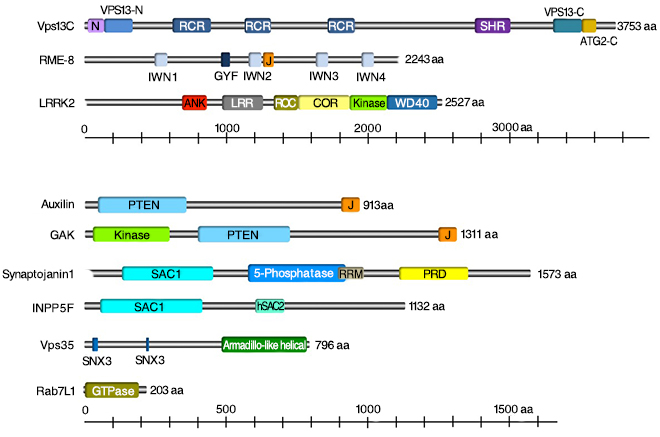
恾9
枌桝憲偵娭傢傞僷乕僉儞僜儞昦堚揱巕嶻暔偺僪儊僀儞峔憿
僪儊僀儞偺徻嵶偼堷梡暥專傪嶲徠偟偰偔偩偝偄乮恾偼丄Inoshita et al., J
Genet. 2018 Figure 4傛傝揮嵹乯丅
|
僔僫僾僗偺僄儞僪僒僀僩乕僔僗
| |
丂恄宱偼僔僫僾僗傪夘偟偰怣崋偺傗傝庢傝傪偟偰偄傑偡乮恾11乯丅僔僫僾僗偼恄宱偲恄宱偁傞偄偼嬝慄堐傗懠偺嵶朎偲偺愙崌晹埵偱偡丅恄宱揱払暔幙傗揹婥怣崋偱怣崋偑傗傝庢傝偝傟傑偡丅恄宱揱払暔幙偼怣崋傪揱偊傞恄宱偐傜彫朎乮摿偵僔僫僾僗彫朎偲屇傇乯偵傛偭偰曻弌偝傟傑偡丅彫朎偼僔僫僾僗枌偲梈崌偟偨屻丄僄儞僪僒僀僩乕僔僗偵傛傝夞廂偝傟傑偡丅僔儑僂僕儑僂僶僄偺恄宱-嬝僔僫僾僗偼僔僫僾僗彫朎偺庢傝崬傒傪傒傞偙偲偵揔偟偰偍傝丄僷乕僉儞僜儞昦尨場堚揱巕偺曄堎偱婲偙傞堎忢傪挷傋偰偄傑偡乮恾12乯丅
恾10
僷乕僉儞僜儞昦堚揱巕嶻暔偑恄宱偱娭傢偭偰偄傞偲峫偊傜傟傞晹埵
彫朎桝憲偵娭傢偭偰偄傞僷乕僉儞僜儞昦尨場堚揱巕偺彮側偔偲傕俀偮埲忋偑丄慜僔僫僾僗偺僄儞僪僒僀僩乕僔僗乮僔僫僾僗彫朎枌偺夞廂乯偲僔僫僾僗彫朎偺嵞惗偵娭傢偭偰偄傞壜擻惈偑憐掕偝傟傞丅堚揱巕娫偺娭學偼丄僔儑僂僕儑僂僶僄暘巕堚揱妛偱柧傜偐偵偱偒傞偲婜懸偝傟傞丅
恾11
僷乕僉儞僜儞昦尨場堚揱巕偵傛傞僔僫僾僗婡擻偺堎忢
僔儑僂僕儑僂僶僄偺恄宱-嬝愙崌晹偺揹婥怣崋傪應掕偟偨僷僞乕儞丅攇宍偵傛傝恄宱偐傜嬝擏傊偺怣崋偑暘偐傞丅僷乕僉儞僜儞昦曄堎偱怣崋偺堎忢乮愒偄揰偱帵偡乯偑尒傜傟傞丅
恾12
僔儑僂僕儑僂僶僄梒拵偺恄宱-嬝愙崌晹埵偺僔僫僾僗
乮嵍乯僔僫僾僗彫朎偼丄傾僋僥傿僽僝乕儞偲屇偽傟傞慜僔僫僾僗枌峔憿偲僪僢僉儞僌偟丄撪晹偵奿擺偟偰偄偨恄宱揱払暔幙傪曻弌偡傞丅
乮塃乯僔僫僾僗偺揹巕尠旝嬀幨恀丅乮忋乯慜僔僫僾僗乮塣摦恄宱廔枛乯傪柍拝怓丄屻僔僫僾僗乮偙偙偱偼嬝嵶朎乯傪惵偱偟傔偡丅M丄儈僩僐儞僪儕傾丅乮壓乯墿怓偺攋慄偺椞堟偺奼戝恾丅傾僋僥傿僽僝乕儞傪栴摢偱帵偡丅僷乕僉儞僜儞昦堚揱巕曄堎偱丄慜僔僫僾僗偵堎忢偵戝偒偄彫朎乮僺儞僋乯偑尒傜傟傞丅彫偝偄彫朎偼丄僔僫僾僗彫朎偱捈宎栺35
nm丅僗働乕儖: 500 nm (忋)丄200 nm乮壓乯丅
僷乕僉儞僜儞昦尨場堚揱巕偼僄儞僪僒僀僩乕僔僗偐傜僔僫僾僗彫朎偺嵞惗傑偱偺夁掱偵娭傢偭偰偄傞偲峫偊傜傟傞丅
|
僷乕僉儞僜儞昦偼僾儕僆儞昦偐丠
| |
丂α-Synuclein偼丄恄宱僔僫僾僗偺奐岥暘斿帪偵僔僫僾僗彫朎傊偺寢崌偲槰棧傪孞傝曉偟偰偄傞偲峫偊傜傟偰偄傑偡乮恾13乯丅僷乕僉儞僜儞昦偱偼丄擼撪偱α-Synuclein偺僑儈乮嬅廤偟慄堐壔偟偨傕偺乯偑棴傑傝丄恄宱曄惈偵宷偑傞偙偲偑柧傜偐偵側傝偮偮偁傝傑偡乮恾14乯丅偙偺α-Synuclein偺僑儈偼丄晠偭偨傒偐傫偺僇價偑懠偺傒偐傫偵揱愼偟偰偄偔傛偆偵丄恄宱夞楬傪捠偠偰擼撪偵峀偑傞偙偲偑幚尡揑偵妋擣偝傟偰偄傑偡乮恾15乯丅偳偺傛偆側偲偒偵恄宱夞楬偵峀偑偭偰偄偔偺偐丄偦偺儕僗僋傪忋徃偝偣傞忦審乮堚揱巕丄恄宱偺妶摦乯偼偳偆偄偭偨傕偺偐?乮恾16乯傪柧傜偐偵偡傞偙偲偑僷乕僉儞僜儞昦偺敪徢傪梊杊偡傞偨傔偵廳梫偱偡丅
丂巹偨偪偼丄α-Synuclein偺僑儈偑偱偒傞僷乕僉儞僜儞昦儌僨儖僴僄傪奐敪偟丄僑儈偑峀偑偭偰偄偔儕僗僋傪忋徃偝偣傞忦審傪柧傜偐偵偟傑偟偨乮恾17乯丅僷乕僉儞僜儞昦尨場堚揱巕PLA2G6/iPLA2β乮昞1嶲徠乯偼丄儕儞帀幙儕僷乕僛傪僐乕僪偟傑偡丅偙偺峺慺偵幘姵偱傒傜傟傞曄堎偑擖傞偲丄壛楊偲嫟偵儕儞帀幙枌偑敄偔側傞偙偲偑暘偐傝傑偟偨丅偦偺寢壥丄α-Synuclein偑寢崌偡傞僔僫僾僗彫朎偺僒僀僘偑彫偝偔側傝丄α-Synuclein偑嵶朎幙偵梀棧偟傗偡偔側傝傑偡丅偙傟偑α-Synuclein偺嬅廤儕僗僋偵側傞偲峫偊傜傟傑偡乮Mori,
PNAS 2019乯丅
丂尰嵼丄α-Synuclein偺僑儈乮昦場峔憿傪傕偪丄嬅廤壔偟偨α-Synuclein乯傪岠棪傛偔専弌偡傞曽朄偑奐敪偝傟偰偄傑偡丅偙偺曽朄傪棙梡偟丄僸僩傗僴僄偱α-Synuclein偺僑儈偑偱偒側偄傛偆偵偡傞愴棯傪扵嶕偟偰偄傑偡乮恾18乯丅
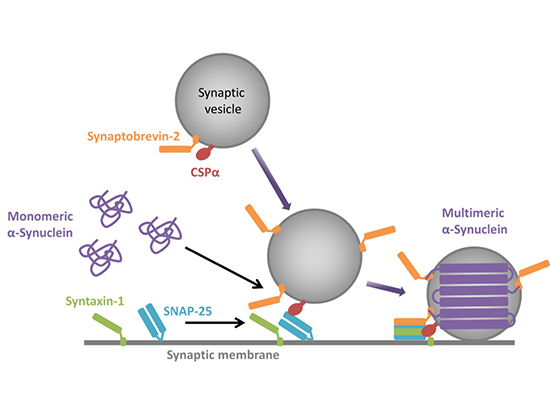
恾13
α-Synuclein偼僔僫僾僗彫朎偺暘斿帪偵懡検壔偲夝懱傪孞傝曉偡
僔僫僾僗彫朎乮Synaptic vesicle乯偑僔僫僾僗枌乮Synaptic
membrane乯偵僪僢僉儞僌偡傞偲偒丄α-Synuclein偼僔僫僾僗彫朎枌忋偱廳崌偡傞(Multimeric
α-Synuclein)丅暘斿偑姰椆偡傞偲丄僔僫僾僗彫朎枌偐傜槰棧偟丄扨検懱偵栠傞(Monomeric
α-Synuclein)丅恄宱妶摦帪偵丄α-Synuclein偼偙偺廳崌偲槰棧傪孞傝曉偡偲峫偊傜傟偰偄傞丅壗傜偐偺偒偭偐偗偱丄α-Synuclein偺峔憿曄壔偑婲偙傝慄堐壔乮僑儈壔乯偡傞乮恾偼丄Inoshita et al., J
Genet. 2018 Figure 5傛傝揮嵹乯丅
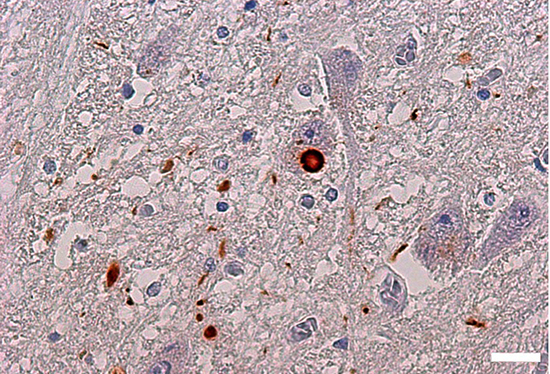
恾14
僷乕僉儞僜儞昦擼偱偺α-Synuclein偺僑儈乮儗價乕彫懱乯
妼怓偱愼傑傞嬅廤懱偑α-Synuclein偺僑儈丅拞墰偺妼怓偺媴懱偑丄恄宱嵶朎懱偵傒傜傟傞儗價乕彫懱偱丄僷乕僉儞僜儞昦偺昦棟恌抐偺儅乕僇乕偺堦偮丅僷乕僉儞僜儞昦擼乮擼嫶乯傪慄堐壔α-Synuclein摿堎揑峈懱偱愼怓偟偨丅僗働乕儖:
20 兪m丅
恾15
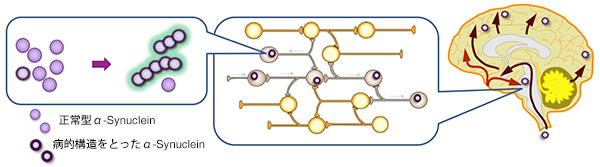
昦揑峔憿傪偲偭偨α-Synuclein偼僾儕僆儞偺傛偆偵揱愼偡傞
恄宱嵶朎撪偱昦揑峔憿曄壔偑惗偠偨α-Synuclein偼丄恄宱娫傪揱斃偟側偑傜昦揑峔憿偺α-Synuclein傪憹暆偡傞丅
恾16
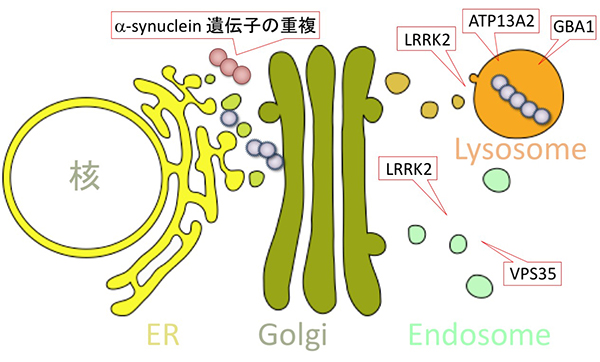
昦揑峔憿偺α-Synuclein偺憹暆偵婑梌偡傞僷乕僉儞僜儞昦尨場堚揱巕
α-Synuclein堚揱巕偺廳暋丗惓忢偱偼1懳乮2僐僺乕乯偺α-Synuclein堚揱巕傪帩偮偑丄堚揱巕偺廳暋偵傛傝丄嶻惗偝傟傞α-Synuclein僞儞僷僋幙偑憹偊傞偙偲偑丄昦場峔憿偺α-Synuclein偺拁愊傗揱斃偵婑梌偡傞丅
嵶朎撪偺桝憲偲僑儈張暘偵娭傢傞堚揱巕偺曄堎丗僷乕僉儞僜儞昦尨場堚揱巕LRRK2,
VPS35,
ATP13A2側偳偼丄僞儞僷僋幙偺桝憲傗僑儈偺暘夝偵娭傢傞偲峫偊傜傟偰偄傞丅偙傟傜偵曄堎偑婲偙傞偙偲偵傛傝丄昦場峔憿偺α-Synuclein偺暘夝偑恑傑偢丄僷乕僉儞僜儞昦傪敪徢偡傞丅乮恾偼丄崱嫃丄僔儕乕僘
傾僋僠儏傾儖 擼丒恄宱幘姵偺椪彴,
僷乕僉儞僜儞昦偲塣摦堎忢丄拞嶳彂揦丂2013傛傝揮嵹乯
恾17
僷乕僉儞僜儞昦尨場堚揱巕PLA2G6偵曄堎偵傛傞α-Synuclein偺嬅廤壔偺儊僇僯僘儉
α-Synuclein偼僔僫僾僗彫朎枌偵寢崌偟婡擻傪敪婗偡傞乮恾13嶲徠乯丅PLA2G6偵曄堎偑偁傞偲僔僫僾僗彫朎偺僒僀僘偑彫偝偔側傞丅偡傞偲枌偑傛傝榩嬋偡傞偨傔丄α-Synuclein偑僔僫僾僗彫朎枌偐傜棧傟傗偡偔側傝丄嬅廤壔偺儕僗僋偵側傞偲峫偊傜傟傞丅
恾18
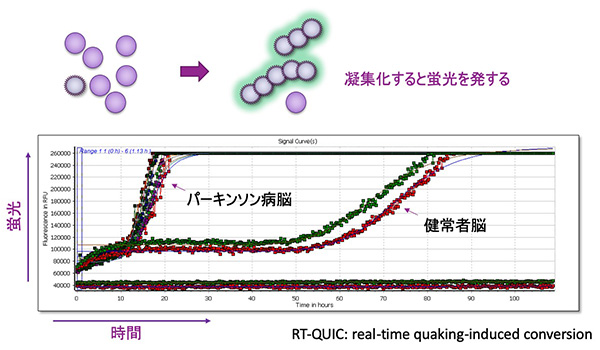
昦場峔憿傪偲偭偨α-Synuclein傪専弌偡傞RT-QUIC朄
昦場峔憿偺α-Synuclein偑丄惓忢側α-Synuclein傪昦場峔憿傊曄姺偟憹暆偡傞惈幙傪棙梡偟丄昦場峔憿偺α-Synuclein偺懚嵼検傪昡壙偡傞丅昦場峔憿偺α-Synuclein偑懡偄偲憗偔寀岝僔僌僫儖偑忋徃偡傞丅
|
儈僩僐儞僪儕傾偲 α-Synuclein
| |
丂僷乕僉儞僜儞昦偱偼儈僩僐儞僪儕傾偺婡擻偑掅壓偟偰偄傞偙偲偑曬崘偝傟偰偄傑偡丅傑偨丄儈僩僐儞僪儕傾偺昳幙娗棟偵娭學偡傞PINK1傗Parkin丄儈僩僐儞僪儕傾屇媧嵔暋崌懱偺揹巕揱払傪惂屼偡傞CHCHD2側偳偑丄僷乕僉儞僜儞昦尨場堚揱巕偲偟偰曬崘偝傟偰偄傑偡丅α-Synuclein偺嬅廤壔傕僷乕僉儞僜儞昦偺敪徢偵宷偑傝傑偡偑丄α-Synuclein偺嬅廤壔偲儈僩僐儞僪儕傾偺娭學偼晄柧偱偟偨丅
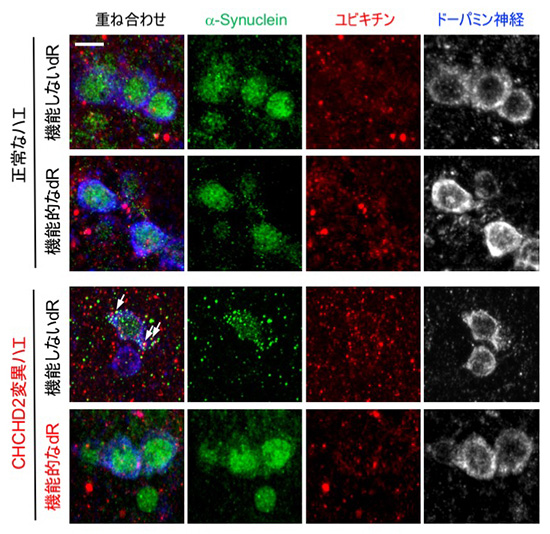
丂巹偨偪偼丄CHCHD2堚揱巕偵曄堎傪傕偭偨僷乕僉儞僜儞昦姵幰偝傫偺擼偵丄α-Synuclein
偺僑儈乮儗價乕彫懱乯偑峀斖埻偵懚嵼偡傞偙偲傪尒偮偗傑偟偨乮恾19乯丅偝傜偵CHCHD2堚揱巕偵曄堎偵傕偭偨姵幰偝傫偐傜嶌惢偟偨iPS嵶朎桼棃偺僪乕僷儈儞恄宱嵶朎丄CHCHD2曄堎堚揱巕傪摫擖偟偨僔儑僂僕儑僂僶僄偱傕丄α-Synuclein
偺僑儈偑棴傑傞偙偲偑暘偐傝傑偟偨乮Ikeda, Hum Mol
Genet. 2019乯丅
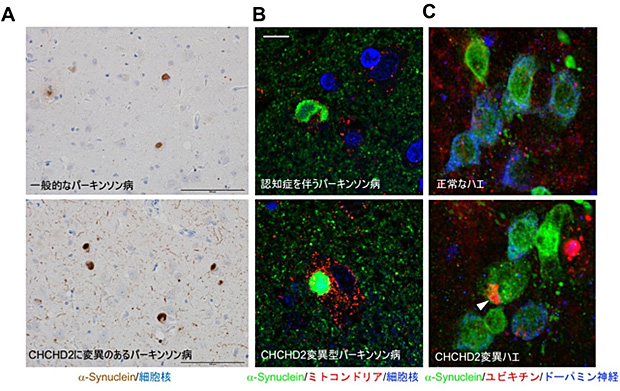
恾19
儈僩僐儞僪儕傾暘巕CHCHD2曄堎偑兛-Synuclein偺嬅廤壔傪摫偔
(A)
CHCHD2偵曄堎偑偁傞僷乕僉儞僜儞昦姵幰偝傫偺擼傪挷傋傞偲丄兛-Synuclein偺拁愊偑峀斖偵尒傜傟偨(壓)丅堦斒揑側僷乕僉儞僜儞昦偺姵幰偝傫(忋)偲斾妑偟偰拁愊偼傛傝尠挊偱偁傞丅娵偄妼怓偺傕偺偑丄儗價乕彫懱乮恾14傕嶲徠乯丅僗働乕儖:
100 兪m. (B)
CHCHD2偵曄堎偑偁傞僷乕僉儞僜儞昦姵幰偝傫(壓)偺儗價乕彫懱偺奼戝乮椢怓偺娵偄峔憿乯丅儈僩僐儞僪儕傾(愒)偼儗價乕彫懱偲偼丄傎偲傫偳堦抳偟側偄丅擣抦徢傪敽偆僷乕僉儞僜儞昦姵幰偝傫乮忋乯偱傕儗價乕彫懱偑峀斖埻偵尒傜傟傞偑丄偦偺儗價乕彫懱偲斾妑偟偰傕戝偒側堘偄偼側偄丅僗働乕儖:
10 兪m.
(C)CHCHD2曄堎傪傕偭偨僴僄偺僪乕僷儈儞恄宱丅兛-Synuclein偺嬅廤乮椢怓偺棻忬偵尒偊傞乯偲儐價僉僠儞乮愒乯偺拁愊乮栴摢乯偑尒偊傞丅暘夝傪扴偆暘巕偱偁傞儐價僉僠儞偼堎忢僞儞僷僋幙偲堦弿偵拁愊偟偰偄傞偲峫偊傜傟傞丅儗價乕彫懱偵傕儐價僉僠儞偑娷傑傟偰偄傞偙偲偑抦傜傟偰偄傞丅
|
儈僩僐儞僪儕傾偵悈慺僀僆儞傪撏偗傞両丠
| |
丂忋婰偱婰偟偨傛偆偵丄CHCHD2堚揱巕偵曄堎偑偁傞偲儈僩僐儞僪儕傾偺婡擻偑掅壓偟丄α-Synuclein
偺僑儈偑棴傑傞偙偲偑暘偐傝傑偟偨乮Ikeda, Hum Mol
Genet.
2019乯丅CHCHD2堚揱巕偵曄堎偑偁傞僴僄偱偼丄儈僩僐儞僪儕傾偐傜懡検偺妶惈巁慺庬偑敪惗偟丄α-Synuclein
偺僑儈傕棴傑傝傑偡乮Meng, Nat
Commun. 2017乯丅
丂悈慺僀僆儞偵偼妶惈巁慺庬傪彍嫀偡傞岠擻偑偁傝傑偡丅偦偙偱巹偨偪偼丄儈僩僐儞僪儕傾偱悈慺僀僆儞傪敪惗偝偣丄妶惈巁慺庬傪彍嫀偡傞偙偲傪帋傒傑偟偨丅徻嵶偵偼丄屆嵶嬠偑傕偮僨儖僞儘僪僾僔儞偲偄偆僞儞僷僋幙傪儈僩僐儞僪儕傾偵摫擖偟傑偟偨乮恾20乯丅偙偺僨儖僞儘僪僾僔儞偼丄岝偑摉偨傞偲悈慺僀僆儞傪塣傃偩偡偲偄偆惈幙傪帩偭偰偄傑偡丅偙偺僨儖僞儘僪僾僔儞偺惈幙傪棙梡偟偰丄岝傪摉偰傞偲儈僩僐儞僪儕傾偺奜懁偵悈慺僀僆儞偑塣傃弌偝傟傞傛偆偵偟傑偟偨丅奜懁偵廤傑偭偨悈慺僀僆儞偼丄妶惈巁慺庬傪彍嫀偟丄偝傜偵儈僩僐儞僪儕傾偺僄僱儖僊乕嶻惗憰抲傪摦偐偟傑偡丅
丂偙偺傾僀僨傾偼惉岟偟丄CHCHD2堚揱巕偵曄堎偑偁傞僷乕僉儞僜儞昦偺儌僨儖僔儑僂僕儑僂僶僄偺儈僩僐儞僪儕傾傪尦婥偵偟丄恄宱曄惈傕梷偊傞偙偲偑偱偒傑偟偨乮Imai,
Commun Biol.
2019乯丅偝傜偵丄嬃偔偙偲偵α-Synuclein
偺僑儈傕棴傑傜側偔側傞偙偲偑暘偐傝傑偟偨乮恾21乯丅偙偺娤嶡偼丄儈僩僐儞僪儕傾偑僞儞僷僋幙偺僑儈傪愊嬌揑偵彍嫀偡傞摥偒偑偁傞偙偲傪帵偟偰偄傑偡丅尰嵼丄偙偺摥偒偺幚懺傪柧傜偐偵偟丄僷乕僉儞僜儞昦偺梊杊朄偵墳梡偡傞偨傔偺尋媶傪恑傔偰偄傑偡丅
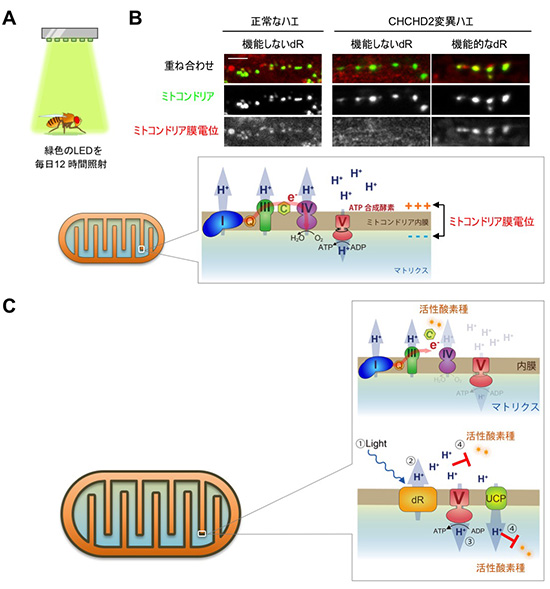
恾20
僨儖僞儘僪僾僔儞偱儈僩僐儞僪儕傾偵悈慺僀僆儞傪撏偗傞
(A)僨儖僞儘僪僾僔儞(dR)傪儈僩僐儞僪儕傾撪枌偵摫擖偟偨CHCHD2曄堎僴僄偵岝傪徠幩偡傞丅僴僄偼丄摢奧崪偑側偄偺偱丄椢怓偺岝偼擼偺怺晹傑偱撏偔丅(B)
dR摫擖偵傛偭偰CHCHD2曄堎僴僄偺儈僩僐儞僪儕傾枌揹埵偑夞暅偟偨丅儈僩僐儞僪儕傾偺枌揹埵偼丄TMRM偲偄偆帋栻偱娤嶡丅岝傪摉偰偰傕婡擻偟側偄dR傪斾妑懳徠偲偟偰抲偄偨丅寬峃側儈僩僐儞僪儕傾偱偼丄怘帠偐傜摼偨摐暘傪嵽椏偵丄屇媧嵔暋崌懱I,
III,
IV偑儅僩儕僋僗偐傜悈慺僀僆儞乮僾儘僩儞乯傪奜偵媯傒弌偡丅師偵媯傒弌偝傟偨僾儘僩儞傪暋崌懱V偑儅僩儕僋僗懁偵栠偡偙偲偵傛傝丄僄僱儖僊乕乮ATP乯偑嶌傜傟傞丅廬偭偰丄寬峃側儈僩僐儞僪儕傾偱偼枌揹埵乮枌傪妘偰偰偺+偲-偺曃傝;
栺-150 mV乯偑堐帩偝傟偰偄傞丅僗働乕儖: 10
兪m.(C)CHCHD2偑夡傟傞偲丄屇媧嵔暋崌懱I, III,
IV傪棳傟傞揹巕偑楻傟丄妶惈巁慺庬偑敪惗偡傞丅揹巕偑楻傟傞偺偱僾儘僩儞傕岠棪傛偔媯傒弌偣偢枌揹埵偑掅壓偡傞乮悂偒弌偟偺忋偺奊乯丅dR偼岝傪摉偰傞偲丄屇媧嵔暋崌懱I,
III,
IV偺曄傢傝偵僾儘僩儞傪媯傒弌偡丅傑偨僾儘僩儞偵偼妶惈巁慺庬傪彍嫀偡傞摥偒偑偁傞丅傛偭偰丄巁壔僗僩儗僗傕梷惂偱偒儈僩僐儞僪儕傾偑寬峃偵曐偨傟傞丅UCP偼儅僩儕僋僗懁偵僾儘僩儞傪栠偟偮偮懱擬傪嶻惗偡傞暘巕丅UCP偱儅僩儕僋僗懁偵栠偭偨僾儘僩儞偼丄儅僩儕僋僗懁偺妶惈巁慺庬傕彍嫀偡傞乮壓偺奊乯丅儈僩僐儞僪儕傾慡懱偺峔憿偼丄恾7傪嶲徠丅
恾21
僨儖僞儘僪僾僔儞偱儈僩僐儞僪儕傾傪寬峃偵偡傞偲丄兛-Synuclein偺嬅廤壔偑慾巭偱偒傞
僨儖僞儘僪僾僔儞(dR)傪摫擖偟偨CHCHD2曄堎僴僄偍傛傃惓忢側僴僄偵岝傪徠幩偟偨丅僾儘僩儞傪媯傒弌偡婡擻傪帩偨側偄dR傪傕偮CHCHD2曄堎僴僄偱偼丄α-Synuclein偺嬅廤壔偑傒傜傟傞乮俁抜栚丄椢怓偺梓棻乯丅偙偺嬅廤偼丄晹暘揑偵儐價僉僠儞偺梓棻忬偺僔僌僫儖(愒)偲堦抳偡傞(栴報)丅婡擻揑側dR傪CHCHD2曄堎僴僄偵摫擖偡傞偲丄α-Synuclein偺嬅廤壔偑梷惂偝傟丄惓忢側僴僄偺傛偆偵側偭偨乮係抜栚乯丅僗働乕儖:
5 兪m.
|
iPS嵶朎偲僴僄儌僨儖傪慻傒崌傢偣偨僷乕僉儞僜儞昦栻偺扵嶕
| |
丂忋婰偱婰偟偨傛偆偵丄儈僩僐儞僪儕傾偺婡擻偑掅壓偡傞偲α-Synuclein
偺僑儈偑棴傑傝傑偡乮Ikeda, Hum Mol
Genet. 2019乯丅媡偵丄儈僩僐儞僪儕傾傪僨儖僞儘僪僾僔儞偱尦婥偵偡傞偲丄α-Synuclein
偺僑儈偑側偔側傞偙偲偑暘偐傝傑偟偨乮Imai, Commun
Biol.
2019乯丅偙偺偙偲偼丄儈僩僐儞僪儕傾傪尦婥偵偡傞栻偑丄僷乕僉儞僜儞昦偺梊杊栻偲偟偰桳朷偱偁傞偙偲傪帵偟偰偄傑偡丅
丂僾儘僥僆儈僋僗夝愅偺崁偱婰嵹偟偨PINK1偲Parkin偼丄儈僩僐儞僪儕傾偺昳幙娗棟傪扴偄傑偡丅偮傑傝婡擻偑掅壓偟偨儈僩僐儞僪儕傾傪彍嫀偟丄寬峃側儈僩僐儞僪儕傾傪巆偡慖暿傪偟偰偄傞偲峫偊傜傟傑偡丅偙傟傜偺堚揱巕偑夡傟傞偲丄庒擭敪徢偺僷乕僉儞僜儞昦偵側傝傑偡丅Parkin偼儐價僉僠儞儕僈乕僛偲偄偆僞儞僷僋幙暘夝偵娭傢傞峺慺偱偡偑丄僷乕僉儞僜儞昦偱偼儈僩僐儞僪儕傾婡擻偑掅壓偟偰傕丄Parkin偑偆傑偔摥偄偰偄側偄偲峫偊傜傟傑偡丅偦偙偱Parkin傪妶惈宆偵偡傞栻傪晲揷栻昳岺嬈偺尋媶幰偲嫤摥偱扵嶕偟丄2偮偺岓曗栻傪尒偮偗傑偟偨乮Shiba-Fukushima, iScience
2020乯丅栻偺昡壙偼丄iPS嵶朎偐傜嶌惢偟偨僪乕僷儈儞恄宱偲PINK1乮Parkin傪妶惈壔偡傞儈僩僐儞僪儕傾偺僉僫乕僛乯偺妶惈偑掅壓偟偨僷乕僉儞僜儞昦儌僨儖僴僄傪巊偄丄昡壙偺掅僐僗僩壔偲僗僺乕僪傾僢僾傪恾傝傑偟偨乮恾22乯丅尰嵼丄尒偮偗偨栻偑儈僩僐儞僪儕傾偵岠偔儊僇僯僘儉偺尋媶傪恑傔偰偄傑偡丅
丂
丂傑偨丄偡偱偵暿偺梡搑偺帯椕栻偲側偭偰偄傞傕偺偺拞偱丄僷乕僉儞僜儞昦姵幰偝傫偺儈僩僐儞僪儕傾傪尦婥偵偡傞傕偺傪丄僷乕僉儞僜儞昦姵幰偝傫偺iPS嵶朎偲僴僄儌僨儖傪慻傒崌傢偣偰昡壙偟丄偦偺惉壥傪敪昞偟偰偄傑偡乮Yamaguchi, Stem
Cell Reports 2020乯丅
恾22
Parkin傪妶惈壔偡傞栻偺扵嶕
(A)儐價僉僠儞儕僈乕僛偱偁傞Parkin偺妶惈傪儖僔僼僃儔乕僛妶惈偱専弌偡傞僗僋儕乕僯儞僌宯傪奐敪偟偨丅4.5枩屄偺掅暘巕壔崌暔傪僗僋儕乕僯儞僌偟丄俀偮偺岓曗傪摨掕偟偨丅(B)僸僩iPS嵶朎偱嶌惢偟偨僪乕僷儈儞恄宱偵偍偄偰丄嵶朎幙偵嬊嵼偡傞Parkin偺儈僩僐儞僪儕傾傊偺堏峴偑栻偺搳梌偱尒傜傟偨乮墿怓偺栴摢乯丅偙傟偼Parkin偑妶惈壔偟偰偄傞偙偲傪帵偟偰偄傞丅僗働乕儖:
10 兪m.
(C,嵍)僔儑僂僕儑僂僶僄嶰楊梒拵偺拞悤擼乮僆儗儞僕乯偺僪乕僷儈儞恄宱乮椢乯偺柾幃恾丅
(C,塃)DL2恄宱妀偺僪乕僷儈儞恄宱乮愒乯偺幨恀丅PINK1偺婡擻偑掅壓偟偨僷乕僉儞僜儞昦儌僨儖乮PINK1儌僨儖僴僄乯偱偼丄Parkin偑摥偐偢儈僩僐儞僪儕傾偺嬅廤偑傒傜傟傞乮墿怓偺栴摢乯丅栻傪搳梌偡傞偲儈僩僐儞僪儕傾偼惓忢側宍懺偵栠傞丅僗働乕儖:
10
兪m.(D)PINK1儌僨儖僴僄梒拵偼儈僩僐儞僪儕傾偺婡擻偑掅壓偟偰摦偒偑埆偄丅栻傪搳梌偡傞偲摦偒偑惓忢偵側傞丅幨恀偼僔儍乕儗偺恀傫拞偵抲偄偨屻丄2暘娫偺梒拵偺堏摦偺婳愓丅
|
崅帀朾怘偑傾儖僣僴僀儅乕昦偺儕僗僋偵側傞儊僇僯僘儉
| |
丂傾儖僣僴僀儅乕昦偼丄兝傾儈儘僀僪偲嬅廤偟偨僞僂偺慄堐偑奀攏偵棴傑傝恄宱曄惈偑婲偙傞昦婥偱偡丅
奀攏偼抁婜婰壇傪拁偊傞偲偙傠偱丄傾儖僣僴僀儅乕昦偱偼偦偺婡擻偑幐傢傟傑偡丅
兝傾儈儘僀僪宍惉偲僞僂偺慄堐壔偺暘巕娭學偼偼偭偒傝暘偐偭偰偄傑偣傫偑丄
僞僂偺慄堐壔偼兝傾儈儘僀僪偺拁愊傛傝屻偵婲偙傞偙偲偑抦傜傟偰偄傑偡丅
慄堐壔偟偨僞僂偼丄僷乕僉儞僜儞昦偱嬅廤偡傞兛-Synuclein摨條丄
僾儕僆儞僞儞僷僋幙偺傛偆偵擼撪偵峀偑偭偰偄偔偙偲偑娤嶡偝傟丄
傾儖僣僴僀儅乕昦偱婲偙傞恄宱曄惈偺幚峴暘巕偲偟偰拲栚偝傟偰偄傑偡丅
丂摐擜昦偑傾儖僣僴僀儅乕昦偺儕僗僋偵側傞偙偲偑塽妛挷嵏偐傜暘偐偭偰偒傑偟偨偑丄
偦偺棟桼偼晄柧偱偟偨丅崅帀朾怘傪怘傋懕偗偨儅僂僗偼摐擜昦偵側傝傑偡偑丄
偦偺嵺敪尰偑曄壔偡傞堚揱巕傪挷嵏偟傑偟偨乮Elahi, Hum
Mol Genet.
2021乯丅
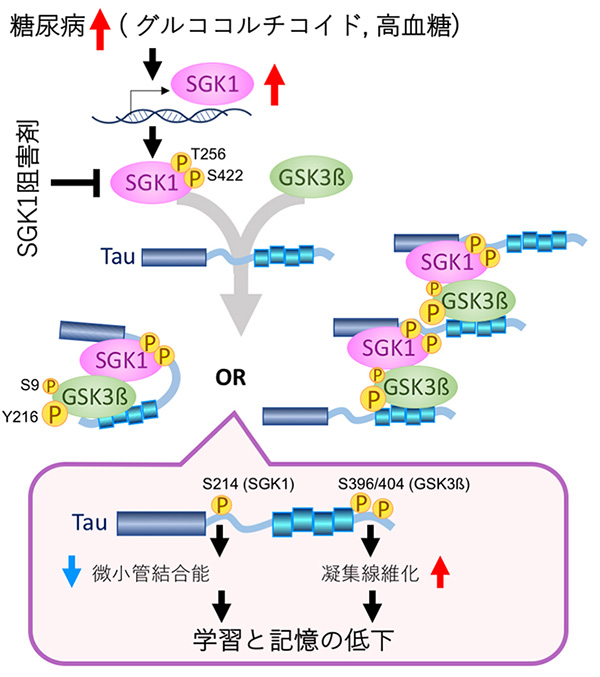
偦偺寢壥丄SGK1偲偄偆僉僫乕僛偺敪尰忋徃偑尒傜傟傑偟偨丅SGK1偼僞僂偺嬅廤壔傪懀恑偡傞GSK3兝偲偄偆僉僫乕僛傪妶惈壔偟丄
偦偺寢壥丄崅帀朾怘傪梌偊偨儅僂僗偺妛廗丒婰壇擻椡偑掅壓偟傑偟偨乮恾23乯丅SGK1偺慾奞嵻傪擼偵搳梌偡傞偲丄偙偺儅僂僗偺妛廗丒婰壇擻椡偑夞暅偟傑偟偨丅
偙偺尋媶偐傜丄SGK1偺慾奞嵻偼傾儖僣僴僀儅乕昦偺栻偲側傞壜擻惈偑峫偊傜傟傑偟偨乮杮尋媶偼杮妛戝妛堾堛妛尋媶壢
擣抦徢恌抐丒梊杊丒帯椕妛島嵗偲偺嫟摨尋媶偱偡乯丅
恾23
傾儖僣僴僀儅乕昦偺尨場偲側傞僞僂偺嬅廤偑摐擜昦偱恑傓儊僇僯僘儉
儅僂僗偵崅帀朾怘傪梌偊懕偗傞偲丄寣塼拞偺僗僩儗僗墳摎儂儖儌儞乮僌儖僐僐儖僠僐僀僪乯偲寣摐偑忋徃偟丄
摐擜昦偺尨場偲側傞僀儞僗儕儞掞峈惈乮僀儞僗儕儞偑岠偒偵偔偔側傞乯傪惗傒弌偡丅
僌儖僐僐儖僠僐僀僪偲崅寣摐偼僞儞僷僋幙僉僫乕僛SGK1偺敪尰傪忋徃偝偣傞丅敪尰偑憹壛偟偨SGK1偼妶惈壔偟丄
僞僂僉僫乕僛GSK3兝傪妶惈壔偡傞偲偲傕偵僞僂偺Ser214傕儕儞巁壔偡傞丅SGK1偵傛傞僞僂偺Ser214偺儕儞巁壔偼丄
僞僂偺旝彫娗寢崌擻偍傛傃旝彫娗埨掕壔擻傪庛傔傞丅旝彫娗偲寢崌偟側偔側偭偨僞僂偼嬅廤丒慄堐壔偟傗偡偔側傞丅
SGK1偱妶惈壔偝傟偨GSK3兝偼丄僞僂偺Ser396/404傪儕儞巁壔偟僞僂傪嬅廤丒慄堐壔偝偣傞丅僞僂偺嬅廤慄堐偼丄奀攏恄宱傪曄惈偝偣丄
妛廗婰壇偺掅壓傪摫偔丅SGK1慾奞嵻偼丄崅帀朾怘傪愛庢偟偨儅僂僗偺妛廗婰壇擻椡偺掅壓傪梷偊傞乮恾偼丄Elahi,
Hum Mol Genet.
2021傛傝堷梡乯丅
|
儈僩僐儞僪儕傾傪娔帇偡傞僷乕僉儞僜儞昦娭楢峺慺
| |
丂Parkin偲PINK1偼丄偲傕偵庒擭惈僷乕僉儞僜儞昦偺尨場堚揱巕偱乮
恾2-4
 亊
丄
昞1
亊
乯丄偙傟傜偺堚揱巕偑摥偐側偔側傞偲10乣40嵨戙偱僷乕僉儞僜儞昦傪敪徢偟傑偡丅PINK1偼儈僩僐儞僪儕傾偵偁傞僞儞僷僋幙儕儞巁壔峺慺偱偁傝丄Parkin偼儐價僉僠儞儕僈乕僛偲偄偆僞儞僷僋幙暘夝峺慺偱偡丅PINK1偼儈僩僐儞僪儕傾偺懝彎傪姶抦偡傞僙儞僒乕偱丄儈僩僐儞僪儕傾偵彎偑偮偔偲Parkin傪儕儞巁壔偟傑偡丅儕儞巁壔偝傟偨Parkin偼丄彎偮偄偨儈僩僐儞僪儕傾偺暘夝偵娭傢傝傑偡丅Parkin堚揱巕傗PINK1堚揱巕偵曄堎偑擖傞偲丄彎偮偄偨儈僩僐儞僪儕傾偺娔帇偑幚峴偝傟側偔側傝丄恄宱嵶朎巰偑婲偙傞偲峫偊傜傟偰偄傑偡丅偟偐偟丄Parkin堚揱巕傗PINK1堚揱巕偑側偄偲丄側偤庒偔偟偰恄宱曄惈偑婲偙傞偺偐偼傑偩暘偐偭偰偄傑偣傫丅
亊
丄
昞1
亊
乯丄偙傟傜偺堚揱巕偑摥偐側偔側傞偲10乣40嵨戙偱僷乕僉儞僜儞昦傪敪徢偟傑偡丅PINK1偼儈僩僐儞僪儕傾偵偁傞僞儞僷僋幙儕儞巁壔峺慺偱偁傝丄Parkin偼儐價僉僠儞儕僈乕僛偲偄偆僞儞僷僋幙暘夝峺慺偱偡丅PINK1偼儈僩僐儞僪儕傾偺懝彎傪姶抦偡傞僙儞僒乕偱丄儈僩僐儞僪儕傾偵彎偑偮偔偲Parkin傪儕儞巁壔偟傑偡丅儕儞巁壔偝傟偨Parkin偼丄彎偮偄偨儈僩僐儞僪儕傾偺暘夝偵娭傢傝傑偡丅Parkin堚揱巕傗PINK1堚揱巕偵曄堎偑擖傞偲丄彎偮偄偨儈僩僐儞僪儕傾偺娔帇偑幚峴偝傟側偔側傝丄恄宱嵶朎巰偑婲偙傞偲峫偊傜傟偰偄傑偡丅偟偐偟丄Parkin堚揱巕傗PINK1堚揱巕偑側偄偲丄側偤庒偔偟偰恄宱曄惈偑婲偙傞偺偐偼傑偩暘偐偭偰偄傑偣傫丅
丂PINK1偑Parkin傪儕儞巁壔偡傞偲丄Parkin偑僞儞僷僋幙暘夝峺慺偲偟偰妶惈壔偡傞偙偲傪曬崘偟傑偟偨乮Shiba-Fukushima,
PLoS Genet. 2014a,
Shiba-Fukushima, PLoS
Genet.
2014b乯乮徻嵶偼偙偪傜傪傒偰偔偩偝偄 乯丅Parkin偑儕儞巁壔傪庴偗傞偲丄Parkin偑儐價僉僠儞偲偄偆彫偝側僞儞僷僋幙傪帺恎偵晅偗傞偙偲傪尒偮偗偰偄傑偟偨偑丄偦偺栶妱偼暘偐傝傑偣傫偱偟偨丅僔儑僂僕儑僂僶僄偱丄Parkin偵儐價僉僠儞偑晅偐側偔側傞曄堎傪摫擖偡傞偲丄Parkin偺妶惈壔岠棪偑掅壓偡傞偙偲偑傢偐傝傑偟偨乮恾24A乯丅徻嵶偵挷傋傞偲丄Parkin偵晅偄偨儐價僉僠儞偼丄PINK1偵傛傝儕儞巁壔偝傟丄Parkin偺妶惈壔傪懀恑偡傞偙偲偑暘偐傝傑偟偨乮恾24B乯丅Parkin偑儐價僉僠儞壔偝傟傞晹埵乮27斣栚偺儕僕儞乯偵曄堎偑偁傞僷乕僉儞僜儞昦姵幰偝傫傕懚嵼偟丄27斣栚偺儕僕儞偺儐價僉僠儞壔偑Parkin偺摥偒偵廳梫偱偁傞偲峫偊傜傟傑偡乮Liu,
Hum Mol Genet.
2022乯丅
乯丅Parkin偑儕儞巁壔傪庴偗傞偲丄Parkin偑儐價僉僠儞偲偄偆彫偝側僞儞僷僋幙傪帺恎偵晅偗傞偙偲傪尒偮偗偰偄傑偟偨偑丄偦偺栶妱偼暘偐傝傑偣傫偱偟偨丅僔儑僂僕儑僂僶僄偱丄Parkin偵儐價僉僠儞偑晅偐側偔側傞曄堎傪摫擖偡傞偲丄Parkin偺妶惈壔岠棪偑掅壓偡傞偙偲偑傢偐傝傑偟偨乮恾24A乯丅徻嵶偵挷傋傞偲丄Parkin偵晅偄偨儐價僉僠儞偼丄PINK1偵傛傝儕儞巁壔偝傟丄Parkin偺妶惈壔傪懀恑偡傞偙偲偑暘偐傝傑偟偨乮恾24B乯丅Parkin偑儐價僉僠儞壔偝傟傞晹埵乮27斣栚偺儕僕儞乯偵曄堎偑偁傞僷乕僉儞僜儞昦姵幰偝傫傕懚嵼偟丄27斣栚偺儕僕儞偺儐價僉僠儞壔偑Parkin偺摥偒偵廳梫偱偁傞偲峫偊傜傟傑偡乮Liu,
Hum Mol Genet.
2022乯丅
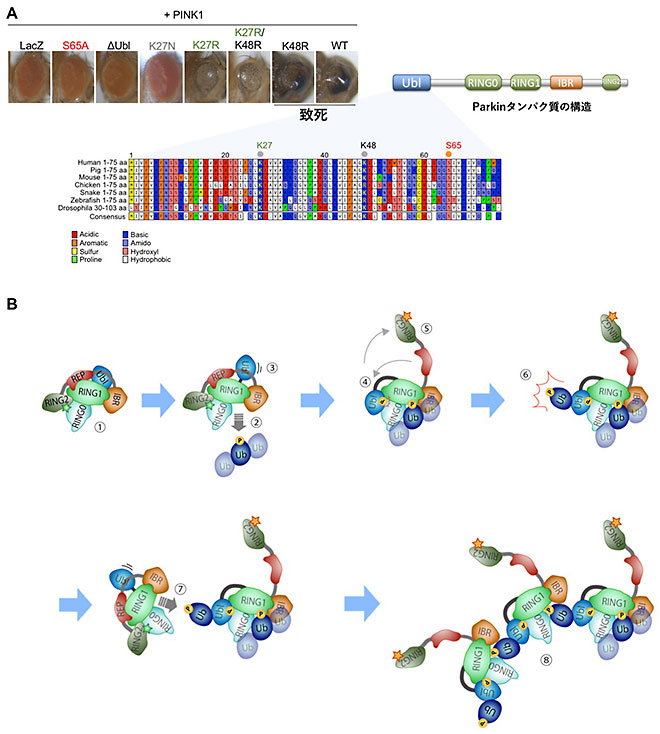
恾24
Parkin偺儕僕儞27斣栚偺儐價僉僠儞壔偑Parkin偺儐價僉僠儞儕僈乕僛妶惈傪憹嫮偡傞
(A乯Parkin偼N枛抂偵儐價僉僠儞條僪儊僀儞乮Ubl, Ubiquitin-like
domain乯傪桳偡傞乮塃恾嶲徠乯丅PINK1偵傛傝65斣栚偺僙儕儞(S65)偑儕儞巁壔偝傟傞偲Parkin偼妶惈壔偟丄儈僩僐儞僪儕傾偺暘夝傪恑傔傞丅惓忢宆乮wild-type,
WT乯偺Parkin傪PINK1偲偲傕偵夁忚敪尰偡傞偲丄栚偺儈僩僐儞僪儕傾偺暘夝偑恑傒丄鍖偺抜奒偱抳巰偵側傞丅堦曽丄PINK1偱儕儞巁壔偝傟側偄S65傪傾儔僯儞偵抲姺偟偨Parkin
(S65A)丄Ubl
傪寚幐偟偨Parkin
(儮Ubl)丄Parkin偲偼娭學側偄僞儞僷僋幙乮LacZ乯偲PINK1偺慻傒崌傢偣偱偼丄栚偼惓忢偵敪惗偡傞丅Parkin偺妶惈壔帪偵儐價僉僠儞壔偝傟傞儕僕儞27(K27)偲K48傪傾儖僊僯儞(R)偵抲姺偟丄儐價僉僠儞壔偝傟側偄傛偆偵偟偨丅偙傟傜Parkin偺偆偪丄K27R曄堎懱偱偼僴僄偑抳巰偵側傜側偐偭偨丅偮傑傝丄Parkin偑儈僩僐儞僪儕傾傪暘夝偡傞妶惈偑庛偄偲峫偊傜傟傞丅僷乕僉儞僜儞昦姵幰偝傫偱尒偮偐偭偨K27N曄堎懱偲PINK1偺慻傒崌傢偣傕丄僴僄偺栚偼惓忢偱偁偭偨丅(B)
K27偺儐價僉僠儞壔傪夘偟偨Parkin偺妶惈壔儌僨儖丅嘆掕忢帪Parkin偼晄妶惈丅嘇儈僩僐儞僪儕傾偵彎偑偮偔偲丄PINK1偑儈僩僐儞僪儕傾忋偺儐價僉僠儞(Ub)嵔傪儕儞巁壔(P)偟丄Parkin偑儕儞巁壔儐價僉僠儞嵔偵寢崌丄峔憿曄姺偑婲偙傞乮嘊乣嘍乯丅嘐峔憿曄姺偵傛傝儐價僉僠儞儕僈乕僛偑妶惈壔偟丄帺恎偺Ubl撪偺K27偵儐價僉僠儞傪晅壛偡傞丅偙偺儐價僉僠儞傪PINK1偑儕儞巁壔(P)偡傞丅嘑嬤朤偵偄傞晄妶惈忬懺偺Parkin偑丄K27偺儕儞巁壔儐價僉僠儞偵寢崌偟妶惈壔丅嘒僗僥僢僾嘐丄嘑偑孞傝曉偝傟丄妶惈壔暋崌懱傪宍惉偡傞丅
|
僷乕僉儞僜儞昦儕僗僋偵側傞兛-Synuclein偺曄堎偲帀幙
| |
丂恄宱偺僔僫僾僗偵偁傞兛-Synuclein乮僷乕僉儞僜儞昦偼僾儕僆儞昦偐丠
傕嶲徠偟偰偔偩偝偄乯偺嬅廤偲丄恄宱夞楬傪夘偟偨僾儕僆儞條偺揱愼乮揱斃乯偼丄僷乕僉儞僜儞昦偺嵟傕堦斒揑側尨場偱偁傞偲峫偊傜傟偰偄傑偡乮懠偺梫場偲偟偰丄儈僩僐儞僪儕傾偑娭學偡傞応崌傕憐掕偝傟偰偄傑偡乯丅兛-Synuclein偼丄僔僫僾僗彫朎偺儕儞帀幙枌偵寢崌偟埨掕壔偡傞乮堦掕偺宍傪偲傞乯偲峫偊傜傟傑偡丅杮朚偱兛-Synuclein堚揱巕偺15斣栚偺僶儕儞偑傾儔僯儞(V15A)偵抲姺偟偨僷乕僉儞僜儞昦壠宯偑2壠宯尒偮偐傝傑偟偨丅V15A偺曄堎偑兛-Synuclein偺惈幙偵曄壔傪傕偨傜偡偐傪挷傋偨偲偙傠丄僔僫僾僗彫朎傪柾曧偟偨儕儞帀幙彫朎偲偺寢崌偑庛偔側傞偙偲丄儕儞帀幙彫朎偐傜梀棧偡傞偲嬅廤偟傗偡偔側傞偙偲傪尒弌偟傑偟偨乮Daida,
Mov Disord.
2022乯丅杮惉壥偼丄柤屆壆戝妛戝妛堾堛妛宯尋媶壢
恄宱撪壢妛嫵幒偲偺嫟摨尋媶偱偡丅
丂忋婰偺娤嶡偐傜兛-Synuclein偼丄僔僫僾僗彫朎偺儕儞帀幙枌傊偺寢崌惈掅壓偑僷乕僉儞僜儞昦偺儕僗僋偵側傞偲峫偊傜傟傑偡乮
恾25
乯丅儕儞帀幙偵寢崌偟偵偔偔側傞梫場偲偟偰丄兛-Synuclein帺恎偺曄堎偲僔僫僾僗彫朎偺帀幙偺慻惉曄壔偑峫偊傜傟傑偡乮
恾17
亊
傕嶲徠偟偰偔偩偝偄乯丅僔僫僾僗彫朎偺帀幙偺慻惉偼丄擔忢偺怘帠偵傛傝彮偟偢偮塭嬁偡傞壜擻惈偑偁傝傑偡丅尰嵼丄偳偺傛偆側帀幙偑僷乕僉儞僜儞昦敪徢偺儕僗僋偺寉尭乮傕偟偔偼憹壛乯偵側傞偺偐尋媶傪恑傔偰偄傑偡丅乽摿掕偺帀幙傪愛庢偟懕偗傟偽丄僷乕僉儞僜儞昦敪徢偺儕僗僋傪梷偊傜傟傞乿偲偄偆偙偲傪幚尰偡傞偙偲傪栚昗偲偟偰偄傑偡丅
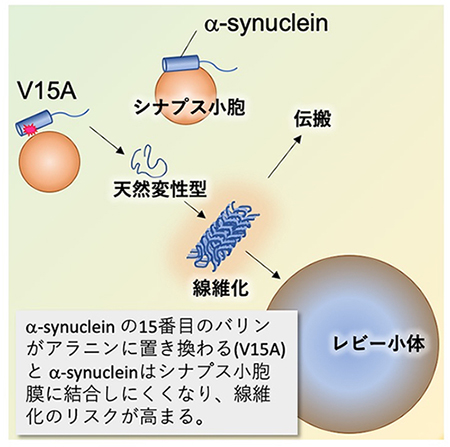
恾25 兛-Synuclein
V15A曄堎偑僷乕僉儞僜儞昦儕僗僋偲側傞暘巕婡彉
V15A曄堎偼丄兛-Synuclein偲僔僫僾僗彫朎偺寢崌傪庛傔傞丅嵶朎幙偵梀棧偟偨兛-Synuclein偼堦掕偺宍傪偲傜側偄偨傔乮揤慠曄惈宆乯丄慄堐壔偺儕僗僋偑崅傑傞丅崅搙偵慄堐壔偟偨兛-Synuclein偑廤傑偭偨傕偺偑丄僷乕僉儞僜儞昦偱傒傜傟傞儗價乕彫懱偺庡惉暘偱偁傞偲峫偊傜傟偰偄傞丅
|
僷乕僉儞僜儞昦儕僗僋偵側傞LRRK2偺曄堎偲兛-Synuclein偲偺娭學
| |
丂LRRK2偼兛-Synuclein偲偲傕偵僷乕僉儞僜儞昦偺儕僗僋堚揱巕偲偟偰廳梫側埵抲傪惉偟偰偄傑偡丅壠懓楌偺側偄乮屒敪惈乯偺僷乕僉儞僜儞昦姵幰偝傫偱傕丄偙傟傜堚揱巕偺曄堎偑偟偽偟偽専弌偝傟傞偐傜偱偡丅LRRK2偼僞儞僷僋幙儕儞巁壔峺慺偱丄暋悢偺僪儊僀儞傪帩偪傑偡乮恾26A,
恾9
亊
傕嶲徠偟偰偔偩偝偄乯丅偙偺偆偪偄偔偮偐偺僪儊僀儞偱昦場曄堎偑悽奅拞偱尒偮偐偭偰偄傑偡丅傑偨丄懡偔偺昦場曄堎偱儕儞巁壔峺慺偺妶惈偑忋徃偟偰偄傑偡丅LRRK2偵曄堎傪傕偮僷乕僉儞僜儞昦姵幰偝傫偺擼偱偼丄兛-Synuclein偺嬅廤偱偁傞儗價乕彫懱偑尒傜傟傞応崌丄尒傜傟側偄応崌丄傾儖僣僴僀儅乕昦側偳偱摿挜揑側儕儞巁壔僞僂偑拁愊偡傞応崌丄側偳偝傑偞傑側昦棟偑傒傜傟傞偙偲偑摿挜偱偡丅
丂LRRK2偺C枛抂偺WD40僪儊僀儞忋偺G2385R曄堎(2385斣栚偺僌儕僔儞巆婎偑傾儖僊僯儞巆婎偵抲偒姺傢傞曄堎)偼寬忢幰傕桳偟傑偡偑丄傾僕傾恖偱偼僷乕僉儞僜儞昦偺儕僗僋偑2攞偵側傞偲曬崘偝傟偰偄傑偡丅巹払偼悽奅偵愭嬱偗偰G2385R曄堎傪傕偮姵幰偝傫偺昦棟夝愅丄惗壔妛揑夝愅傪恑傔傑偟偨乮Tezuka,
NPJ Parkinsons Dis
2022乯丅偦偺寢壥丄G2385R曄堎傪傕偮擼偱偼儕儞巁壔峺慺妶惈偑忋徃偟偰偍傝丄儗價乕彫懱偺拁愊丄儕儞巁壔僞僂偺拁愊丄椉曽偑傒傜傟傑偟偨乮恾26B乯丅堦曽丄LRRK2偺儕儞巁壔峺慺妶惈偑崅偄晹埵偲儗價乕彫懱偺拁愊晹埵偵偼憡娭偑偁傝傑偣傫偱偟偨乮恾26C乯丅傑偨LRRK2偼擼偺墛徢偵娭梌偡傞壜擻惈偑峫偊傜傟偰偄傑偡偑丄墛徢偺僒僀儞偱偁傞傾僗僩儘僒僀僩傗儈僋儘僌儕傾偺妶惈壔偼拞掱搙偱偟偨乮恾26B乯丅杮夝愅偺寢壥偐傜丄LRRK2偺儕儞巁壔峺慺偺忋徃偼兛-Synuclein偺嬅廤傗揱斃偵捈愙娭傢傞偲偄偆傛傝偼丄傓偟傠擼偺榁壔傪恑傔傞栶妱傪傕偭偰偄傞偲峫偊傜傟傑偟偨丅
丂LRRK2偺儕儞巁壔峺慺妶惈偺忋徃偑僷乕僉儞僜儞昦偺尨場偱偁傞僪乕僷儈儞恄宱曄惈傪傕偨傜偡偙偲偐傜丄悽奅拞偱LRRK2偺慾奞嵻偑奐敪偝傟偰偄傑偡丅偟偐偟丄LRRK2偼攛傗恡憻偱傕廳梫側栶妱傪偟偰偄傞偙偲偑暘偐偭偰偍傝丄姰慡偵慾奞偡傞偲攛傗恡憻偺婡擻偵塭嬁偡傞偙偲偑梊憐偝傟傑偡丅LRRK2偺擼偱偺栶妱偺夝柧偲LRRK2偺峺慺妶惈偺揔搙側挷愡偑丄僷乕僉儞僜儞昦崕暈偺堦偮偺壽戣偵側偭偰偄傑偡丅
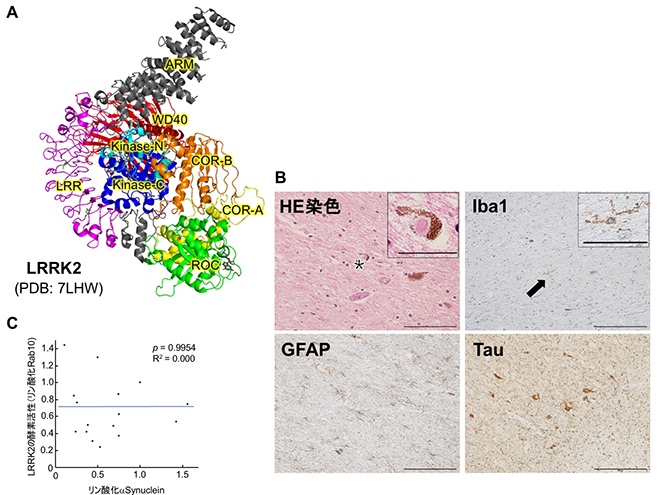
恾26 傾僕傾恖偵懡偄LRRK2
G2385R曄堎偵偮偄偰偺悽奅弶偺擼昦棟曬崘
(A)掅壏揹巕尠旝嬀朄偱悇掕偝傟傞LRRK2偺3師尦峔憿乮抈敀幙峔憿僨乕僞僶儞僋
偺僨乕僞傛傝昤夋乯丅WD40偼儕儞巁壔峺慺僪儊僀儞偺堦晹乮Kinase-N乯傪暍偆宍偱攝埵偟偰偄傞丅G2385R偼WD40偺峔憿傪晄埨掕偟丄儕儞巁壔峺慺僪儊僀儞偵傕塭嬁偡傞偲梊憐偝傟傞丅
(B)LRRK2
G2385R傪桳偡傞堦徢椺偼揟宆揑側儗價乕彫懱昦棟傪帵偡丅僿儅僩僉僔儕儞丒僄僆僕儞(HE)愼怓偵偰丄揟宆揑側儗價乕彫懱偑娤嶡偝傟傞乮憓擖偼*偺椞堟偺奼戝幨恀乯丅儈僋儘僌儕傾乮憓擖偼栴報偺椞堟偺奼戝幨恀乯偲傾僗僩儘僒僀僩偼丄偦傟偧傟Iba1偲GFAP偺峈懱偱愼怓丅儕儞巁壔僞僂(Tau)傕尠挊偵拁愊偟偰偄傞丅僗働乕儖僶乕:
HE愼怓, 100 µm; Iba1, GFAP, Tau, 200
µm; 奼戝幨恀, 25 µm丅
(C)LRRK2偺儕儞巁壔峺慺妶惈偲兛-Synuclein偺嬅廤偵偼憡娭偑側偄丅僌儔僼偺墶幉偼兛-Synuclein偺嬅廤乮儗價乕彫懱乯傪尒愊傕傞儕儞巁壔兛-Synuclein偺奺擼椞堟偺掕検丅廲幉偼丄LRRK2偺峺慺妶惈偺巜昗偱偁傞儕儞巁壔Rab10偺奺擼椞堟偺掕検丅
|
僷乕僉儞僜儞昦尨場堚揱巕CHCHD2偲嬝堔弅惈懁嶕峝壔徢(ALS)偺娭學
| |
丂僷乕僉儞僜儞昦尨場堚揱巕偲偟偰摨掕偝傟偨CHCHD2偼丄僠僩僋儘儉C傪儈僩僐儞僪儕傾屇媧嵔暋崌懱偵埨掕壔偝偣丄揹巕揱払傪惂屼偟傑偡乮僷乕僉儞僜儞昦尨場堚揱巕偵傛傞儈僩僐儞僪儕傾婡擻偺惂屼丂偺崁傕嶲徠偟偰偔偩偝偄乯丅堦曽偱丄ALS傗慜摢懁摢宆擣抦徢(FTD)偺尨場堚揱巕偲偟偰CHCHD10偑曬崘偝傟偰偄傑偡丅CHCHD10偲CHCHD2偼憃巕偺娭學偱偡丅偮傑傝恑壔揑偵堦偮偺堚揱巕偐傜俀偮偺僐僺乕偵暘偐傟偨偲峫偊傜傟丄椉暘巕偺傾儈僲巁攝楍偼椶帡偟偰偄傑偡丅巹払偼丄CHCHD2偺曄堎傕ALS偺儕僗僋偵側傞壜擻惈傪峫偊丄ALS姵幰偝傫偺僎僲儉DNA傪挷傋傑偟偨(Ikeda, PNAS Nexus 2024)丅偦偺寢壥丄14斣栚偺僾儘儕儞偑儘僀僔儞偵傾儈僲巁抲姺乮P14L乯偟偨曄堎傪傕偮ALS姵幰偝傫傪尒偮偗傑偟偨丅揟宆揑側ALS偱偼丄TDP-43偲偄偆RNA寢崌僞儞僷僋幙偑塣摦恄宱偵嬅廤丒拁愊偟傑偡偑丄CHCHD2 P14L傪傕偭偨姵幰偝傫偵傕嬅廤壔偟偨TDP-43偑棴傑偭偰偄傑偟偨丅CHCHD2偼儈僩僐儞僪儕傾枌娫峯偵懚嵼偟丄憃巕暘巕CHCHD10偲暋崌懱傪宍惉偟偰偄傑偡丅偟偐偟P14L曄堎偵傛偭偰丄CHCHD10偲偺寢崌偑庛偔側傝丄儈僩僐儞僪儕傾偐傜嵶朎幙偵楻傟傗偡偔側傞偙偲偑暘偐傝傑偟偨丅偦偺寢壥丄儈僩僐儞僪儕傾偺Ca2+娚徴婡擻偑掅壓偟丄TDP-43偺愗抐偲嬅廤壔偑恑傓偙偲傪柧傜偐偵偟傑偟偨乮徻嵶偼丄恾27偲Ikeda, PNAS Nexus 2024傪嶲徠偟偰壓偝偄乯丅
丂偙偺尋媶偱偼丄屒敪惈乮壠懓楌偑側偄乯ALS姵幰偝傫偱CHCHD2偲CHCHD10偺mRNA偺敪尰忋徃傪娤嶡偟傑偟偨丅儈僩僐儞僪儕傾偵僗僩儗僗偑偐偐傞偲CHCHD2傗CHCHD10偼敪尰忋徃偡傞偙偲偐傜丄屒敪惈ALS姵幰偝傫偱傕儈僩僐儞僪儕傾忈奞偑婲偙偭偰偄傞偙偲偑帵嵈偝傟傑偡丅嬝堔弅惈懁嶕峝壔徢丒僷乕僉儞僜儞丒擣抦徢暋崌乮Kii ALS/PDC乯偲偄偆幘姵偑偁傝傑偡丅婭埳敿搰偱偟偽偟偽尒傜傟傞晽搚昦偱偡偑丄堚揱揑梫場傗娐嫬梫場偼埶慠偲偟偰晄柧偱偡丅Kii ALS/PDC偺姵幰偝傫iPS嵶朎偐傜暘壔偝偣偨傾僗僩儘僒僀僩偱偼丄CHCHD2偺敪尰掅壓偲儈僩僐儞僪儕傾婡擻晄慡偑曬崘偝傟偰偄傑偡(PMID: 38750212乯丅偙偺傛偆偵丄CHCHD2偼僷乕僉儞僜儞昦偲ALS憃曽偺昦懺偵娭梌偡傞壜擻惈偑帵嵈偝傟傑偡丅
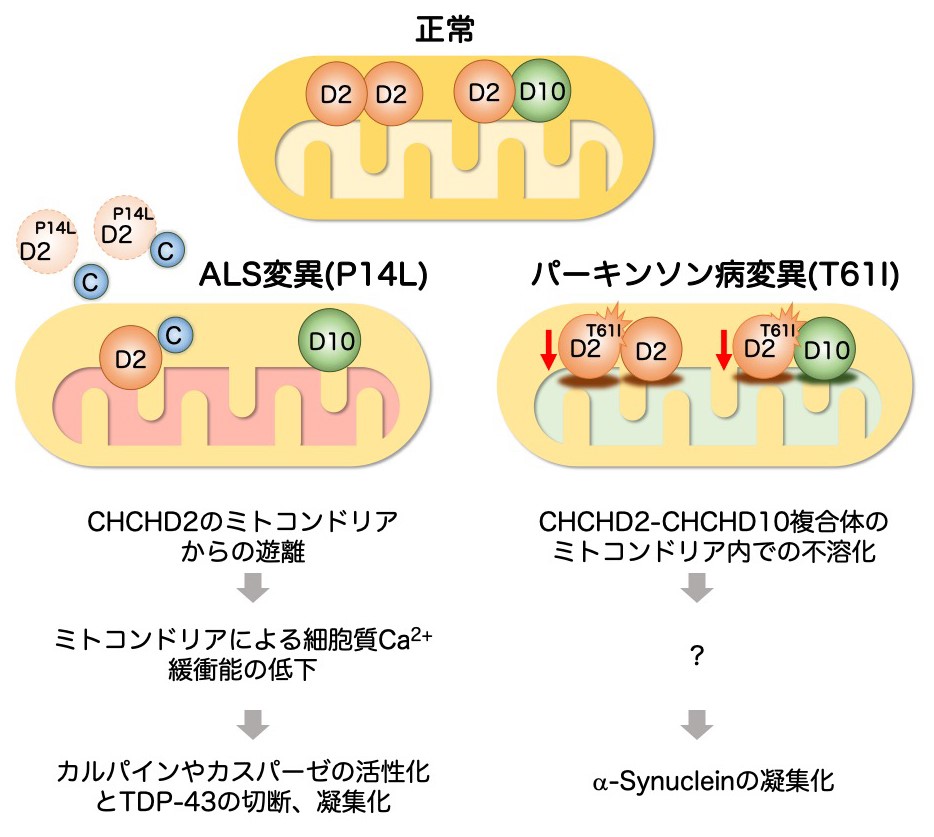
恾27 CHCHD2曄堎偑ALS偲僷乕僉儞僜儞昦傪堷偒婲偙偡暘巕婡彉乮壖愢乯
乮嵍乯塣摦恄宱偼恄宱妶摦偵敽偄嵶朎幙Ca2+偑崅傑傞偑丄帩懕揑偵Ca2+擹搙偑崅偄忬懺偑懕偔偲恄宱撆惈偵偮側偑傞丅彫朎懱傗儈僩僐儞僪儕傾偼丄崅偔側偭偨嵶朎幙Ca2+傪庢傝崬傓嶌梡乮娚徴嶌梡乯傪傕偮丅ALS偱専弌偝傟偨P14L曄堎偼丄儈僩僐儞僪儕傾偐傜嵶朎幙傊偺CHCHD2偺梀棧傪懀恑偡傞丅偦偺寢壥丄儈僩僐儞僪儕傾偵傛傞嵶朎幙Ca2+娚徴擻偑掅壓偡傞丅嵶朎幙Ca2+偑崅偄忬懺偑懕偔偲丄儈僩僐儞僪儕傾偐傜偝傜偵CHCHD2傗僠僩僋儘儉C (C)偑梀棧偡傞丅梀棧偟偨僠僩僋儘儉C偼僇僗僷乕僛傪妶惈壔偡傞丅傑偨崅擹搙偺嵶朎幙Ca2+偼僇儖僷僀儞傪妶惈壔偡傞丅僇僗僷乕僛傗僇儖僷僀儞偼TDP-43傪愗抐偟丄C枛抂懁偺揤慠曄惈晹埵偺嬅廤壔傪懀恑偡傞丅乮塃乯僷乕僉儞僜儞昦偱傒傜傟傞T61I曄堎偼丄CHCHD2偲偦偺寢崌僷乕僩僫乕CHCHD10偺晄梟壔傪傕偨傜偡丅晄梟壔偟偨CHCHD2-CHCHD10偼儈僩僐儞僪儕傾僗僩儗僗傪堷偒婲偙偟丄兛-Synuclein偺嬅廤壔傪懀恑偡傞丅
|
|
|
